Hybrid lipid bioelectronic membranes
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Hybrid solid-state chips and biological cells integrated at molecular level
http://www.kurzweilai.net/hybrid-solid-state-chips-and-biological-cells-integrated-at-molecular-level

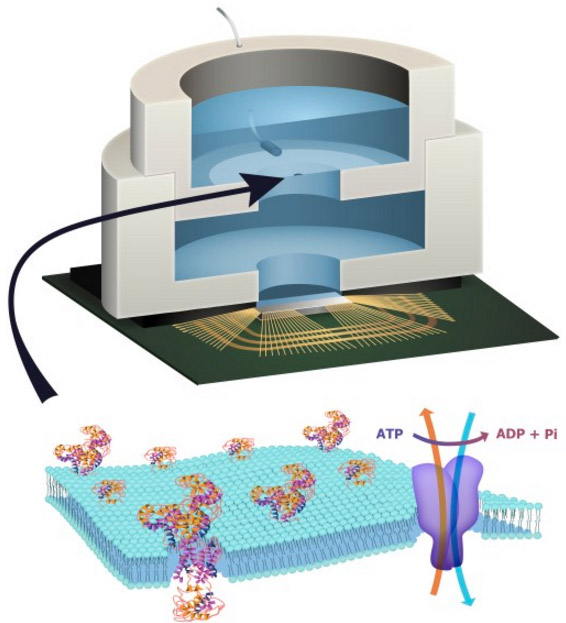
Columbia Engineering researchers have combined biological and solid-state components for the first time, opening the door to creating entirely new artificial biosystems.
In this experiment, they used a biological cell to power a conventional solid-state complementary metal-oxide-semiconductor (CMOS) integrated circuit. An artificial lipid bilayer membrane containing adenosine triphosphate (ATP)-powered ion pumps (which provide energy for cells) was used as a source of ions (which were converted to electrons to power the chip).
The study, led by Ken Shepard, Lau Family Professor of Electrical Engineering and professor of biomedical engineering at Columbia Engineering, was published online today (Dec. 7, 2015) in an open-access paper in Nature Communications.
How to build a hybrid biochip
Living systems achieve this functionality with their own version of electronics based on lipid membranes and ion channels and pumps, which act as a kind of “biological transistor.” Charge in the form of ions carry energy and information, and ion channels control the flow of ions across cell membranes.
Solid-state systems, such as those in computers and communication devices, use electrons; their electronic signaling and power are controlled by field-effect transistors.
To build a prototype of their hybrid system, Shepard’s team packaged a CMOS integrated circuit (IC) with an ATP-harvesting “biocell.” In the presence of ATP, the system pumped ions across the membrane, producing an electrical potential (voltage)* that was harvested by the integrated circuit.
“We made a macroscale version of this system, at the scale of several millimeters, to see if it worked,” Shepard notes. “Our results provide new insight into a generalized circuit model, enabling us to determine the conditions to maximize the efficiency of harnessing chemical energy through the action of these ion pumps. We will now be looking at how to scale the system down.”
While other groups have harvested energy from living systems, Shepard and his team are exploring how to do this at the molecular level, isolating just the desired function and interfacing this with electronics. “We don’t need the whole cell,” he explains. “We just grab the component of the cell that’s doing what we want. For this project, we isolated the ATPases because they were the proteins that allowed us to extract energy from ATP.”
The capability of a bomb-sniffing dog, no Alpo required
Next, the researchers plan to go much further, such as recognizing specific molecules and giving chips the potential to taste and smell.
The ability to build a system that combines the power of solid-state electronics with the capabilities of biological components has great promise, they believe. “You need a bomb-sniffing dog now, but if you can take just the part of the dog that is useful — the molecules that are doing the sensing — we wouldn’t need the whole animal,” says Shepard.
The technology could also provide a power source for implanted electronic devices in ATP-rich environments such as inside living cells, the researchers suggest.
* “In general, integrated circuits, even when operated at the point of minimum energy in subthreshold, consume on the order of 10−2 W mm−2 (or assuming a typical silicon chip thickness of 250 μm, 4 × 10−2 W mm−3). Typical cells, in contrast, consume on the order of 4 × 10−6 W mm−3. In the experiment, a typical active power dissipation for the IC circuit was 92.3 nW, and the active average harvesting power was 71.4 fW for the biocell (the discrepancy is managed through duty-cycled operation of the IC).” — Jared M. Roseman et al./Nature Communications
Hybrid integrated biological–solid-state system powered with adenosine triphosphate
Jared M. Roseman, Jianxun Lin, Siddharth Ramakrishnan, Jacob K. Rosenstein & Kenneth L. Shepard
Nature Communications 7 Dec 2015; 6(10070) http://dx.doi.org:/10.1038/ncomms10070
There is enormous potential in combining the capabilities of the biological and the solid state to create hybrid engineered systems. While there have been recent efforts to harness power from naturally occurring potentials in living systems in plants and animals to power complementary metal-oxide-semiconductor integrated circuits, here we report the first successful effort to isolate the energetics of an electrogenic ion pump in an engineered in vitro environment to power such an artificial system. An integrated circuit is powered by adenosine triphosphate through the action of Na+/K+ adenosine triphosphatases in an integrated in vitro lipid bilayer membrane. The ion pumps (active in the membrane at numbers exceeding 2 × 106 mm−2) are able to sustain a short-circuit current of 32.6 pA mm−2 and an open-circuit voltage of 78 mV, providing for a maximum power transfer of 1.27 pW mm−2 from a single bilayer. Two series-stacked bilayers provide a voltage sufficient to operate an integrated circuit with a conversion efficiency of chemical to electrical energy of 14.9%.
Figure 1: Fully hybrid biological–solid-state system.

http://www.nature.com/ncomms/2015/151207/ncomms10070/images/ncomms10070-f1.jpg
(a) Illustration depicting biocell attached to CMOS integrated circuit. (b) Illustration of membrane in pore containing sodium–potassium pumps. (c) Circuit model of equivalent stacked membranes,  =2.1 pA,
=2.1 pA,  =98.6 GΩ,
=98.6 GΩ,  =575 GΩ and
=575 GΩ and  =75 pF, Ag/AgCl electrode equivalent resistance RWE+RCE<20 kΩ, energy-harvesting capacitor CSTOR=100 nF combined with switch as an impedance transformation network (only one switch necessary due to small duty cycle), and CMOS IC voltage doubler and resistor representing digital switching load. RL represents the four independent ring oscillator loads. (d) Equivalent circuit detail of stacked biocell. (e) Switched-capacitor voltage doubler circuit schematic.
=75 pF, Ag/AgCl electrode equivalent resistance RWE+RCE<20 kΩ, energy-harvesting capacitor CSTOR=100 nF combined with switch as an impedance transformation network (only one switch necessary due to small duty cycle), and CMOS IC voltage doubler and resistor representing digital switching load. RL represents the four independent ring oscillator loads. (d) Equivalent circuit detail of stacked biocell. (e) Switched-capacitor voltage doubler circuit schematic.
The energetics of living systems are based on electrochemical membrane potentials that are present in cell plasma membranes, the inner membrane of mitochondria, or the thylakoid membrane of chloroplasts1. In the latter two cases, the specific membrane potential is known as the proton-motive force and is used by proton adenosine triphosphate (ATP) synthases to produce ATP. In the former case, Na+/K+-ATPases hydrolyse ATP to maintain the resting potential in most cells.
While there have been recent efforts to harness power from some naturally occurring potentials in living systems that are the result of ion pump action both in plants2 and animals3, 4 to power complementary metal-oxide semiconductor (CMOS) integrated circuits (ICs), this work is the first successful effort to isolate the energetics of an electrogenic ion pump in an engineered in vitroenvironment to power such an artificial system. Prior efforts to harness power from in vitromembrane systems incorporating ion-pumping ATPases5, 6, 7, 8, 9 and light-activated bacteriorhodopsin9, 10, 11 have been limited by difficulty in incorporating these proteins in sufficient quantity to attain measurable current and in achieving sufficiently large membrane resistances to harness these currents. Both problems are solved in this effort to power an IC from ATP in an in vitro environment. The resulting measurements provide new insight into a generalized circuit model, which allows us to determine the conditions to maximize the efficiency of harnessing chemical energy through the action of electrogenic ion pumps.
ATP-powered IC
Figure 1a shows the complete hybrid integrated system, consisting of a CMOS IC packaged with an ATP-harvesting ‘biocell’. The biocell consists of two series-stacked ATPase bearing suspended lipid bilayers with a fluid chamber directly on top of the IC. Series stacking of two membranes is necessary to provide the required start-up voltage for IC and eliminates the need for an external energy source, which is typically required to start circuits from low-voltage supplies2, 3. As shown inFig. 1c, a matching network in the form of a switched capacitor allows the load resistance of the IC to be matched to that presented by the biocell. In principle, the switch S can be implicit. The biocell charges CSTOR until the self start-up voltage, Vstart, is reached. The chip then operates until the biocell voltage drops below the minimum supply voltage for operation, Vmin. Active current draw from the IC stops at this point, allowing the charge to build up again on CSTOR. In our case, however, the IC leakage current exceeds 13.5 nA at Vstart, more than can be provided by the biocell. As a result, an explicit transistor switch and comparator (outside of the IC) are used for this function in the experimental results presented here, which are not powered by the biocell and not included in energy efficiency calculations (see Supplementary Discussion for additional details). The energy from the biocell is used to operate a voltage converter (voltage doubler) and some simple inverter-based ring oscillators in the IC, which receive power from no other sources.
http://www.nature.com/ncomms/2015/151207/ncomms10070/images/ncomms10070-f1.jpg
…….. Prior to the addition of ATP, the membrane produces no electrical power and has an Rm of 280 GΩ. A 1.7-pA short-circuit (SC) current (Fig. 2b) through the membrane is observed upon the addition of ATP (final concentration 3 mM) to the cis chamber where functional, properly oriented enzymes generate a net electrogenic pump current. To perform these measurements, currents through each membrane of the biocell are measured using a voltage-clamp amplifier (inset of Fig. 2b) with a gain of 500 GΩ with special efforts taken to compensate amplifier leakage currents. Each ATPase transports three Na+ ions from the cis chamber to the trans chamber and two K+ ions from thetrans chamber to the cis chamber (a net charge movement of one cation) for every molecule of ATP hydrolysed. At a rate of 100 hydrolysis events per second under zero electrical (SC) bias13, this results in an electrogenic current of ~16 aA. The observed SC current corresponds to about 105 active ATPases in the membrane or a concentration of about 2 × 106 mm−2, about 5% of the density of channels occurring naturally in mammalian nerve fibres14. It is expected that half of the channels inserted are inactive because they are oriented incorrectly.
Figure 2: Single-cell biocell characterization.
http://www.nature.com/ncomms/2015/151207/ncomms10070/images_article/ncomms10070-f2.jpg
(a)…Pre-ATP data linear fit (black line) slope yield Rm=280 GΩ. Post ATP data fit to a Boltzmann curve, slope=0.02 V (blue line). Post-ATP linear fit (red line) yields Ip=−1.8 pA and Rp=61.6 GΩ, which corresponds to a per-ATP source resistance of 6.16 × 1015. The current due to membrane leakage through R_{m} is subtracted in the post-ATP curve…. (b)…
Current–voltage characteristics of the ATPases
Figure 2a shows the complete measured current–voltage (I–V) characteristic of a single ATPase-bearing membrane in the presence of ATP. The current due to membrane leakage through Rm is subtracted in the post-ATP curve. The I–V characteristic fits a Boltzmann sigmoid curve, consistent with sodium–potassium pump currents measured on membrane patches at similar buffer conditions13, 15, 16. This nonlinear behaviour reflects the fact that the full ATPase transport cycle (three Na+ ions from cis to trans and two K+ ions from trans to cis) time increases (the turn-over rate, kATP, decreases) as the membrane potential increases16. No effect on pump current is expected from any ion concentration gradients produced by the action of the ATPases (seeSupplementary Discussion). Using this Boltzmann fit, we can model the biocell as a nonlinear voltage-controlled current source IATPase (inset Fig. 2a), in which the current produced by this source varies as a function of Vm. In the fourth quadrant, where the cell is producing electrical power, this model can be linearized as a Norton equivalent circuit, consisting of a DC current source (Ip) in parallel with a current-limiting resistor (Rp), which acts to limit the current delivered to the load at increasing bias (IATPase~Ip−Vm/Rp). Figure 2c shows the measured and simulated charging of Cm for a single membrane (open-circuited voltage). A custom amplifier with input resistance Rin>10 TΩ was required for this measurement (see Electrical Measurement Methods).
Reconciling operating voltage differences
The electrical characteristics of biological systems and solid-state systems are mismatched in their operating voltages. The minimum operating voltage of solid-state systems is determined by the need for transistors to modulate a Maxwell–Boltzmann (MB) distribution of carriers by several orders of magnitude through the application of a potential that is several multiples of kT/q (where kis Boltzmann’s constant, T is the temperature in degrees Kelvin and q is the elementary charge). Biological systems, while operating under the same MB statistics, have no such constraints for operating ion channels since they are controlled by mechanical (or other conformational) processes rather than through modulation of a potential barrier. To bridge this operating voltage mismatch, the circuit includes a switched-capacitor voltage doubler (Fig. 1d) that is capable of self-startup from voltages as low Vstart=145 mV (~5.5 kT/q) and can be operated continuously from input voltages from as low as Vmin=110 mV (see Supplementary Discussion)…..
Maximizing the efficiency of harvesting energy from ATP
Solid-state systems and biological systems are also mismatched in their operating impedances. In our case, the biocell presents a source impedance,  =84.2 GΩ, while the load impedance presented by the complete integrated circuit (including both the voltage converter and ring oscillator loads) is approximately RIC=200 kΩ. (The load impedance, RL, of the ring oscillators alone is 305 kΩ.) This mismatch in source and load impedance is manifest in large differences in power densities. In general, integrated circuits, even when operated at the point of minimum energy in subthreshold, consume on the order of 10−2 W mm−2 (or assuming a typical silicon chip thickness of 250 μm, 4 × 10−2 W mm−3) (ref. 17). Typical cells, in contrast, consume on the order of 4 × 10−6 W mm−3 (ref. 18). In our case, a typical active power dissipation for our circuit is 92.3 nW, and the active average harvesting power is 71.4 fW for the biocell. This discrepancy is managed through duty-cycled operation of the IC in which the circuit is largely disabled for long periods of time (Tcharge), integrating up the power onto a storage capacitor (CSTOR), which is then expended in a very brief period of activity (Trun), as shown in Fig. 3a.
=84.2 GΩ, while the load impedance presented by the complete integrated circuit (including both the voltage converter and ring oscillator loads) is approximately RIC=200 kΩ. (The load impedance, RL, of the ring oscillators alone is 305 kΩ.) This mismatch in source and load impedance is manifest in large differences in power densities. In general, integrated circuits, even when operated at the point of minimum energy in subthreshold, consume on the order of 10−2 W mm−2 (or assuming a typical silicon chip thickness of 250 μm, 4 × 10−2 W mm−3) (ref. 17). Typical cells, in contrast, consume on the order of 4 × 10−6 W mm−3 (ref. 18). In our case, a typical active power dissipation for our circuit is 92.3 nW, and the active average harvesting power is 71.4 fW for the biocell. This discrepancy is managed through duty-cycled operation of the IC in which the circuit is largely disabled for long periods of time (Tcharge), integrating up the power onto a storage capacitor (CSTOR), which is then expended in a very brief period of activity (Trun), as shown in Fig. 3a.
The overall efficiency of the system in converting chemical energy to the energy consumed in the load ring oscillator (η) is given by the product of the conversion efficiency of the voltage doubler (ηconverter) and the conversion efficiency of chemical energy to electrical energy in the biocell (ηbiocell), η=ηconverter × ηbiocell. ηconverter is relatively constant over the range of input voltages at ~59%, as determined by various loading test circuits included in the chip design (Supplementary Figs 1–6). ηbiocell, however, varies with transmembrane potential Vm. η is the efficiency in transferring power to the power ring oscillator loads from the ATP harvested by biocell.
…….
To first order, the energy made available to the Na+/K+-ATPase by the hydrolysis of ATP is independent of the chemical or electric potential of the membrane and is given by |ΔGATP|/(qNA), where ΔGATP is the Gibbs free energy change due to the ATP hydrolysis reaction per mole of ATP at given buffer conditions and NA is Avogadro’s number. Since every charge that passes through IATPase corresponds to a single hydrolysis event, we can use two voltage sources in series with IATPase to independently account for the energy expended by the pumps both in moving charge across the electric potential difference and in moving ions across the chemical potential difference. The dependent voltage source Vloss in this branch fixes the voltage across IATPase, and the total power produced by the pump current source is (|ΔGATP|/NA)(NkATP), which is the product of the energy released per molecule of ATP, the number of active ATPases and the ATP turnover rate. The power dissipated in voltage source Vchem models the work performed by the ATPases in transporting ions against a concentration gradient. In the case of the Na+/K+ ATPase,Vchem is given by  . The power dissipated in this source is introduced back into the circuit in the power generated by the Nernst independent voltage sources,
. The power dissipated in this source is introduced back into the circuit in the power generated by the Nernst independent voltage sources,  and
and  . The power dissipated in the dependent voltage source Vloss models any additional power not used to perform chemical or electrical work. ……
. The power dissipated in the dependent voltage source Vloss models any additional power not used to perform chemical or electrical work. ……
Integration of ATP-harvesting ion pumps could provide a means to power future CMOS microsystems scaled to the level of individual cells22. In molecular diagnostics, the integration of pore-forming proteins such as alpha haemolysin23 or MspA porin24 with CMOS electronics is already finding application in DNA sequencing25. Exploiting the large diversity of function available in transmembrane proteins in these hybrid systems could, for example, lead to highly specific sensing platforms for airborne odorants or soluble molecular entities26, 27. Heavily multiplexed platforms could become high-throughput in vitro drug-screening platforms against this diversity of function. In addition, integration of transmembrane proteins with CMOS may become a convenient alternative to fluorescence for coupling to synthetic biological systems28.
Roseman, J. M. et al. Hybrid integrated biological–solid-state system powered with adenosine triphosphate. Nat. Commun. 6:10070 http://dx.doi.org:/10.1038/ncomms10070 (2015).
- The measurement of membrane potential and deltapH in cells, organelles, and vesicles. Methods Enzymol. 55, 547–569 (1979).
- , , , & Ultralow voltage nanoelectronics powered directly, and solely, from a tree. IEEE Trans. Nanotechnol. 9, 2–5(2010).
- , , , & Energy extraction from the biologic battery in the inner ear. Nat. Biotechnol. 30, 1240–1243(2012).
- et al. Implanted Biofuel Cell Operating in a Living Snail. J. Am. Chem. Soc.134, 5040–5043 (2012).


 Thursday 15 October 2015, 12:00–13:00
Thursday 15 October 2015, 12:00–13:00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}