Telling NO to Cardiac Risk
DDAH Says NO to ADMA(1); The DDAH/ADMA/NOS Pathway(2)
Author-Writer-Reporter: Stephen J. Williams, PhD
Endothelium-derived nitric oxide (NO) has been shown to be vasoprotective. Nitric oxide enhances endothelial cell survival, inhibits excessive proliferation of vascular smooth muscle cells, regulates vascular smooth muscle tone, and prevents platelets from sticking to the endothelial wall. Together with evidence from preclinical and human studies, it is clear that impairment of the NOS pathway increases risk of cardiovascular disease (3-5).
This post contains two articles on the physiological regulation of nitric oxide (NO) by an endogenous NO synthase inhibitor asymmetrical dimethylarginine (ADMA) and ADMA metabolism by the enzyme DDAH(1,2). Previous posts on nitric oxide, referenced at the bottom of the page, provides excellent background and further insight for this posting. In summary plasma ADMA levels are elevated in patients with cardiovascular disease and several large studies have shown that plasma ADMA is an independent biomarker for cardiovascular-related morbidity and mortality(6-8).
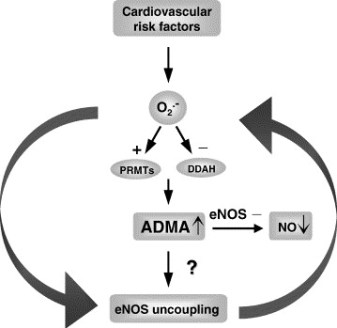

Figure 1 A. Cardiac risks of ADMA B. Effects of ADMA (Photo credit: Wikipedia)
ADMA Production and Metabolism
Nuclear proteins such as histones can be methylated on arginine residues by protein-arginine methyltransferases, enzymes which use S-adenosylmethionine as methyl groups. This methylation event is thought to regulate protein function, much in the way of protein acetylation and phosphorylation (9). And much like phosphorylation, these modifications are reversible through methylesterases. The proteolysis of these arginine-methyl modifications lead to the liberation of free guanidine-methylated arginine residues such as L-NMMA, asymmetric dimethylarginine (ADMA) and symmetrical methylarginine (SDMA).
The first two, L-NMMA and ADMA, have been shown to inhibit the activity of the endothelial NOS. This protein turnover is substantial: for instance the authors note that each day 40% of constitutive protein in adult liver is newly synthesized protein. And in several diseases, such as muscular dystrophy, ischemic heart disease, and diabetes, it has been known since the 1970’s that protein catabolism rates are very high, with corresponding increased urinary excretion of ADMA(10-13). Methylarginines are excreted in the urine by cationic transport. However, the majority of ADMA and L-NMMA are degraded within the cell by dimethylaminohydrolase (DDAH), first cloned and purified in rat(14).

Figure 2. Endogenous inhibitors of NO synthase. Chemical structures generated from PubChem.
DDAH
DDAH specifically hydrolyzes ADMA and L-NMMA to yield citruline and demethylamine and usually shows co-localization with NOS. Pharmacologic inhibition of DDAH activity causes accumulation of ADMA and can reverse the NO-mediated bradykinin-induced relaxation of human saphenous vein.
Two isoforms have been found in human:
- DDAH1 (found in brain and kidney and associated with nNOS) and
- DDAH2 (highly expressed in heart, placenta, and kidney and associated with eNOS).
DDAH2 can be upregulated by all-trans retinoic acid (atRA can increase NO production). Increased reactive oxygen species and possibly homocysteine, a risk factor for cardiovascular disease, can decrease DDAH activity(15,16).
- The importance of DDAH activity can also be seen in transgenic mice which overexpress DDAH, exhibiting increased NO production, increased insulin sensitivity, and reduced vascular resistance (17). Likewise,
- Transgenic mice, null for the DDAH1, showed increase in blood pressure, decreased NO production, and significant increase in tissue and plasma ADMA and L-NMMA.

Figure 3. The DDAH/ADMA/NOS cycle. Figure adapted from Cooke and Ghebremarian (1).
As mentioned in the article by Cooke and Ghebremariam, the authors state: the weight of the evidence indicates that DDAH is a worthy therapeutic target. Agents that increase DDAH expression are known, and 1 of these, a farnesoid X receptor agonist, is in clinical trials
http://www.interceptpharma.com
An alternate approach is to
- develop an allosteric activator of the enzyme. Although
- development of an allosteric activator is not a typical pharmaceutical approach, recent studies indicate that this may be achievable aim(18).
References:
1. Cooke, J. P., and Ghebremariam, Y. T. : DDAH says NO to ADMA.(2011) Arteriosclerosis, thrombosis, and vascular biology 31, 1462-1464
2. Tran, C. T., Leiper, J. M., and Vallance, P. : The DDAH/ADMA/NOS pathway.(2003) Atherosclerosis. Supplements 4, 33-40
3. Niebauer, J., Maxwell, A. J., Lin, P. S., Wang, D., Tsao, P. S., and Cooke, J. P.: NOS inhibition accelerates atherogenesis: reversal by exercise. (2003) American journal of physiology. Heart and circulatory physiology 285, H535-540
4. Miyazaki, H., Matsuoka, H., Cooke, J. P., Usui, M., Ueda, S., Okuda, S., and Imaizumi, T. : Endogenous nitric oxide synthase inhibitor: a novel marker of atherosclerosis.(1999) Circulation 99, 1141-1146
5. Wilson, A. M., Shin, D. S., Weatherby, C., Harada, R. K., Ng, M. K., Nair, N., Kielstein, J., and Cooke, J. P. (2010): Asymmetric dimethylarginine correlates with measures of disease severity, major adverse cardiovascular events and all-cause mortality in patients with peripheral arterial disease. Vasc Med 15, 267-274
6. Kielstein, J. T., Impraim, B., Simmel, S., Bode-Boger, S. M., Tsikas, D., Frolich, J. C., Hoeper, M. M., Haller, H., and Fliser, D. : Cardiovascular effects of systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans.(2004) Circulation 109, 172-177
7. Kielstein, J. T., Donnerstag, F., Gasper, S., Menne, J., Kielstein, A., Martens-Lobenhoffer, J., Scalera, F., Cooke, J. P., Fliser, D., and Bode-Boger, S. M. : ADMA increases arterial stiffness and decreases cerebral blood flow in humans.(2006) Stroke; a journal of cerebral circulation 37, 2024-2029
8. Mittermayer, F., Krzyzanowska, K., Exner, M., Mlekusch, W., Amighi, J., Sabeti, S., Minar, E., Muller, M., Wolzt, M., and Schillinger, M. : Asymmetric dimethylarginine predicts major adverse cardiovascular events in patients with advanced peripheral artery disease.(2006) Arteriosclerosis, thrombosis, and vascular biology 26, 2536-2540
9. Kakimoto, Y., and Akazawa, S.: Isolation and identification of N-G,N-G- and N-G,N’-G-dimethyl-arginine, N-epsilon-mono-, di-, and trimethyllysine, and glucosylgalactosyl- and galactosyl-delta-hydroxylysine from human urine. (1970) The Journal of biological chemistry 245, 5751-5758
10. Inoue, R., Miyake, M., Kanazawa, A., Sato, M., and Kakimoto, Y.: Decrease of 3-methylhistidine and increase of NG,NG-dimethylarginine in the urine of patients with muscular dystrophy. (1979) Metabolism: clinical and experimental 28, 801-804
11. Millward, D. J.: Protein turnover in skeletal muscle. II. The effect of starvation and a protein-free diet on the synthesis and catabolism of skeletal muscle proteins in comparison to liver. (1970) Clinical science 39, 591-603
12. Goldberg, A. L., and St John, A. C.: Intracellular protein degradation in mammalian and bacterial cells: Part 2. (1976) Annual review of biochemistry 45, 747-803
13. Dice, J. F., and Walker, C. D.: Protein degradation in metabolic and nutritional disorders. (1979) Ciba Foundation symposium, 331-350
14. Ogawa, T., Kimoto, M., and Sasaoka, K.: Purification and properties of a new enzyme, NG,NG-dimethylarginine dimethylaminohydrolase, from rat kidney. (1989) The Journal of biological chemistry 264, 10205-10209
15. Ito, A., Tsao, P. S., Adimoolam, S., Kimoto, M., Ogawa, T., and Cooke, J. P.: Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. (1999) Circulation 99, 3092-3095
16. Stuhlinger, M. C., Tsao, P. S., Her, J. H., Kimoto, M., Balint, R. F., and Cooke, J. P. : Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine.(2001) Circulation 104, 2569-2575
17. Sydow, K., Mondon, C. E., Schrader, J., Konishi, H., and Cooke, J. P.: Dimethylarginine dimethylaminohydrolase overexpression enhances insulin sensitivity. (2008) Arteriosclerosis, thrombosis, and vascular biology 28, 692-697
18. Zorn, J. A., and Wells, J. A.: Turning enzymes ON with small molecules. (2010) Nature chemical biology 6, 179-188
Other research papers on Nitric Oxide and Cardiac Risk were published on this Scientific Web site as follows:
The Nitric Oxide and Renal is presented in FOUR parts:
Part I: The Amazing Structure and Adaptive Functioning of the Kidneys: Nitric Oxide
Part II: Nitric Oxide and iNOS have Key Roles in Kidney Diseases
Part III: The Molecular Biology of Renal Disorders: Nitric Oxide
Part IV: New Insights on Nitric Oxide donors
Cardiac Arrhythmias: A Risk for Extreme Performance Athletes
What is the role of plasma viscosity in hemostasis and vascular disease risk?
Biochemistry of the Coagulation Cascade and Platelet Aggregation – Part I
Dr. Williams,
THIS IS OUTSTANDING, seminal work at it’s best with the biochemistry centrality of the ETIOLOGY of Cardiac Risk and its comorbidities in context of the major cause of mortality in the Western World.
I am primarily pleased with the outcome of my own decision to commission YOU to take upon yourself the research and authoring involved in this post.
This post supplements a long list of seminal scientific writings and reporting on the Frontiers in Cardiology, Frontiers in the Etiology of Cardiovascular Diseases and Innovations in Cardiac Repair: valve repair in TAVI, Stents With Nitric Oxide sustained release coatings.
This post will be included in our Nitric Oxide e-Book and in our Cardiovascular e-Book, both have a forthcoming release in 2013.
Very proud and honored for your decision to join our Team and to make significant contributions in several domains, having your Senior Editorial talent harnessed for the Cancer e-Book, you guiding another three Editors for a body of publications exceeding 150 existing posts. The e-Book on Cancer is targeted for all Attending Oncologists Worldwide. And you are a gifted Expert, Author, Writer (EAW) on Nitric Oxide, on Cardivascular Disase, and will have contributions to the PharmacoGenomic e-book in 2014.
Thank you again for captivating my mind and my imagination with your scholarship and being always ready to work on my new commissions. As explained, they mirror my recognition of EAWs’ capacity to innovate.
We are running a tight ship, I see, the lines of a bright seashore ahead for this equity sharing venture, we all contribute to, to the best of our abilities.
Steve, Great post. Thanks for following up on the ADMA pathway. Here is another reference Böger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the ‘L-arginine paradox’ and acts as a novel cardiovascular risk factor. J Nutr 2004; 134: 2842S–7S http://www.ncbi.nlm.nih.gov/pubmed/15465797
Dr. Williams, This is a great post. Thank you!!! I actually wanted to post something in the next couple of days regarding NO and cardiothoratic surgery. I will re-adjust my post to accommodate it to yours.
Thank you, Tilda
Thank you Aviva, Meg, and Tilda! And a special thanks to Aviva. This is a real exciting topic, something I knew nothing about but this area seems to be exploding and a great target for cardiovascular disease. I think, Meg that would be a great post (on the L-arginine paradox) and I was surprised to see another paper on ADMA in nociception and opiate dependency. Looking for your post Dr. Barliya on cardiothoracic surgery which would have a great clinical aspect for the NO posts. Everyone here does exemplary work and I am very happy to be associated with all of you.
Who, in this World, can imagine the pure joy I have, to have assembled all of you for great synergy and the advancement of the scientific discourse. And be the way, we are to create economic value along the way, sooner than later.
Dr williams what an amazing post.Congratulations!You talk about of usefullness of ADMA.If i’m giving quemo(QT) to a patient with an anthraciclin in the protocol,do you think it could have a place to monitor cardiac toxicity?Or help to decide if the patient cannot have doxo or epirrubicin on ADMA basis?
[1] plasma ADMA levels are elevated in patients with cardiovascular disease and several large studies have shown that plasma ADMA is an independent biomarker for cardiovascular-related morbidity and mortality
[2] Nuclear proteins such as histones can be methylated on arginine residues by protein-arginine methyltransferases, enzymes which use S-adenosylmethionine as methyl groups. This methylation event is thought to regulate protein function, much in the way of protein acetylation and phosphorylation
This is very interesting. Homocysteine is an independent predictor of CVD. In other words, unrelated to the lipid profiles. But it has only been used in patients with CVD who lack the more prevalent risk factors. I might infer from this report that ADMA is correlated with homocysteinemia, and that neither is independent.
Why is this? The key is unavailability of S-adenosylmethionine for the methylation process. Whether this is independent of phosphorylations I cannot say, but the energy of the cell depends on ~P in Coenzyme A. The TCA cycle depends on Acetyl CoA.
Take a step back and look at the methylation issue. There are 3 key methyl donors: cobalamine (B-12), folic acid, A-adenosyl methionine. Cobalamine has relevance for anemia and neuropathy, but it has to my knowledge, no relevance to CAD. Yet there are individuals who are thin and develop CAD and experience acute myocardial infarction (also diabetes). In this case S-adenosyl methionine is suspect, which would result in homocysteinemia.
Why is this? The total body sulfur is critical, as is nitrogen. Sulfur is available in field that have been deposited by the rich lava of volcanos. There are sparse, arid lands that are sulfur poor. Plants have a S:N ratio of 1:20, while animals have a S:N ratio of 1:12.5. This has been accurately measured and reported by the now deceased Vernon Young (MIT), and Yves Ingenbleek (Univ Louis Pasteur). Ingenbleek showed that in the plains of Africa compaired to the nearby mountain community, the plains people had a nearly vegan diet, having no access to animal protein. This study was done with the distinguished Harvard professor who discovered the relationship of plaque to hyperhomocysteinemia.
The deficiency in dietary sulfur protein, thus, is associated with hyperhomocystenemia and an increased CVD rate. Ingenbleek went on to establish that transthyretin is the best measure of protein loss either by protein energy malnutrition or by stress hypermetabolism. In both cases, the body has to tear down lean body mass (proteolysis) for gluconeogenic precursors. This ties in with the process you describe. In this case there would be both low TTR and elevated homocysteine. I am not aware of whether in diabetes, itself a risk factor with insulin resistance, there is also hyperhomocysteinemia.
Legumes are high in sulfer content (includes beans and jicama), as are plants in the allium family (onions and garlic) and cruciferous vegetables and kale. Egg yolks are very good. So not all about animal protein in the diet. Ethnobotanical studies usually shows that diets of a culture evolve to include good sources of necessary nutrients. Western diets have lost contact with this and we have crazy notions causing people to exclude things like egg yolks from their diets.
I actually consider this amazing blog , âSAME SCIENTIFIC IMPACT: Scientific Publishing –
Open Journals vs. Subscription-based « Pharmaceutical Intelligenceâ, very compelling plus the blog post ended up being a good read.
Many thanks,Annette