Colon cancer and organoids
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Guts and Glory
An open mind and collaborative spirit have taken Hans Clevers on a journey from medicine to developmental biology, gastroenterology, cancer, and stem cells.
Ihave had to talk a lot about my science recently and it’s made me think about how science works,” says Hans Clevers. “Scientists are trained to think science is driven by hypotheses, but for [my lab], hypothesis-driven research has never worked. Instead, it has been about trying to be as open-minded as possible—which is not natural for our brains,” adds the Utrecht University molecular genetics professor. “The human mind is such that it tries to prove it’s right, so pursuing a hypothesis can result in disaster. My advice to my own team and others is to not preformulate an answer to a scientific question, but just observe and never be afraid of the unknown. What has worked well for us is to keep an open mind and do the experiments. And find a collaborator if it is outside our niche.”
“One thing I have learned is that hypothesis-driven research tends not to be productive when you are in an unknown territory.”
Clevers entered medical school at Utrecht University in The Netherlands in 1978 while simultaneously pursuing a master’s degree in biology. Drawn to working with people in the clinic, Clevers had a training position in pediatrics lined up after medical school, but then mentors persuaded him to spend an additional year converting the master’s degree to a PhD in immunology. “At the end of that year, looking back, I got more satisfaction from the research than from seeing patients.” Clevers also had an aptitude for benchwork, publishing four papers from his PhD year. “They were all projects I had made up myself. The department didn’t do the kind of research I was doing,” he says. “Now that I look back, it’s surprising that an inexperienced PhD student could come up with a project and publish independently.”
Clevers studied T- and B-cell signaling; he set up assays to visualize calcium ion flux and demonstrated that the ions act as messengers to activate human B cells, signaling through antibodies on the cell surface. “As soon as the experiment worked, I got T cells from the lab next door and did the same experiment. That was my strategy: as soon as something worked, I would apply it elsewhere and didn’t stop just because I was a B-cell biologist and not a T-cell biologist. What I learned then, that I have continued to benefit from, is that a lot of scientists tend to adhere to a niche. They cling to these niches and are not that flexible. You think scientists are, but really most are not.”
Here, Clevers talks about promoting a collaborative spirit in research, the art of doing a pilot experiment, and growing miniature organs in a dish.
Clevers Creates
Re-search? Clevers was born in Eindhoven, in the south of The Netherlands. The town was headquarters to Philips Electronics, where his father worked as a businessman, and his mother took care of Clevers and his three brothers. Clevers did well in school but his passion was sports, especially tennis and field hockey, “a big thing in Holland.” Then in 1975, at age 18, he moved to Utrecht University, where he entered an intensive, biology-focused program. “I knew I wanted to be a biology researcher since I was young. In Dutch, the word for research is ‘onderzoek’ and I knew the English word ‘research’ and had wondered why there was the ‘re’ in the word, because I wanted to search but I didn’t want to do re-search—to find what someone else had already found.”
Opportunity to travel. “I was very disappointed in my biology studies, which were old-fashioned and descriptive,” says Clevers. He thought medicine might be more interesting and enrolled in medical school while still pursuing a master’s degree in biology at Utrecht. For the master’s, Clevers had to do three rotations. He spent a year at the International Laboratory for Research on Animal Diseases (ILRAD) in Nairobi, Kenya, and six months in Bethesda, Maryland, at the National Institutes of Health. “Holland is really small, so everyone travels.” Clevers saw those two rotations more as travel explorations. In Nairobi, he went on safaris and explored the country in Land Rovers borrowed from the institute. While in Maryland in 1980, Clevers—with the consent of his advisor, who thought it was a good idea for him to get a feel for the U.S.—flew to Portland, Oregon, and drove back to Boston with a musician friend along the Canadian border. He met the fiancé of political activist and academic Angela Davis in New York City and even stayed in their empty apartment there.
Life and lab lessons. Back in Holland, Clevers joined Rudolf Eugène Ballieux’s lab at Utrecht University to pursue his PhD, for which he studied immune cell signaling. “I didn’t learn much science from him, but I learned that you always have to create trust and to trust people around you. This became a major theme in my own lab. We don’t distrust journals or reviewers or collaborators. We trust everyone and we share. There will be people who take advantage, but there have only been a few of those. So I learned from Ballieux to give everyone maximum trust and then change this strategy only if they fail that trust. We collaborate easily because we give out everything and we also easily get reagents and tools that we may need. It’s been valuable to me in my career. And it is fun!”
Clevers Concentrates
On a mission. “Once I decided to become a scientist, I knew I needed to train seriously. Up to that point, I was totally self-trained.” From an extensive reading of the immunology literature, Clevers became interested in how T cells recognize antigens, and headed off to spend a postdoc studying the problem in Cox Terhorst’s lab at Dana-Farber Cancer Institute in Boston. “Immunology was young, but it was very exciting and there was a lot to discover. I became a professional scientist there and experienced how tough science is.” In 1988, Clevers cloned and characterized the gene for a component of the T-cell receptor (TCR) called CD3-epsilon, which binds antigen and activates intracellular signaling pathways.
On the fast track in Holland. Clevers returned to Utrecht University in 1989 as a professor of immunology. Within one month of setting up his lab, he had two graduate students and a technician, and the lab had cloned the first T cell–specific transcription factor, which they called TCF-1, in human T cells. When his former thesis advisor retired, Clevers was asked, at age 33, to become head of the immunology department. While the appointment was high-risk for him and for the department, Clevers says, he was chosen because he was good at multitasking and because he got along well with everyone.
Problem-solving strategy. “My strategy in research has always been opportunistic. One thing I have learned is that hypothesis-driven research tends not to be productive when you are in an unknown territory. I think there is an art to doing pilot experiments. So we have always just set up systems in which something happens and then you try and try things until a pattern appears and maybe you formulate a small hypothesis. But as soon as it turns out not to be exactly right, you abandon it. It’s a very open-minded type of research where you question whether what you are seeing is a real phenomenon without spending a year on doing all of the proper controls.”
Trial and error. Clevers’s lab found that while TCF-1 bound to DNA, it did not alter gene expression, despite the researchers’ tinkering with promoter and enhancer assays. “For about five years this was a problem. My first PhD students were leaving and they thought the whole TCF project was a failure,” says Clevers. His lab meanwhile cloned TCF homologs from several model organisms and made many reagents including antibodies against these homologs. To try to figure out the function of TCF-1, the lab performed a two-hybrid screen and identified components of the Wnt signaling pathway as binding partners of TCF-1. “We started to read about Wnt and realized that you study Wnt not in T cells but in frogs and flies, so we rapidly transformed into a developmental biology lab. We showed that we held the key for a major issue in developmental biology, the final protein in the Wnt cascade: TCF-1 binds b-catenin when b-catenin becomes available and activates transcription.” In 1996, Clevers published the mechanism of how the TCF-1 homolog in Xenopus embryos, called XTcf-3, is integrated into the Wnt signaling pathway.
Clevers Catapults

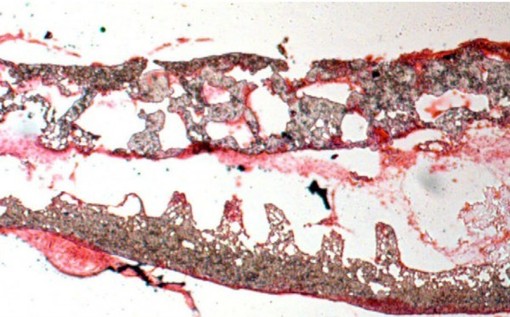
COURTESY OF HANS CLEVERS AND JEROEN HUIJBEN, NYMUS
3DCrypt building and colon cancer.
Clevers next collaborated with Bert Vogelstein’s lab at Johns Hopkins, linking TCF to Wnt signaling in colon cancer. In colon cancer cell lines with mutated forms of the tumor suppressor gene APC, the APC protein can’t rein in b-catenin, which accumulates in the cytoplasm, forms a complex with TCF-4 (later renamed TCF7L2) in the nucleus, and caninitiate colon cancer by changing gene expression. Then, the lab showed that Wnt signaling is necessary for self-renewal of adult stem cells, as mice missing TCF-4 do not have intestinal crypts, the site in the gut where stem cells reside. “This was the first time Wnt was shown to play a role in adults, not just during development, and to be crucial for adult stem cell maintenance,” says Clevers. “Then, when I started thinking about studying the gut, I realized it was by far the best way to study stem cells. And I also realized that almost no one in the world was studying the healthy gut. Almost everyone who researched the gut was studying a disease.” The main advantages of the murine model are rapid cell turnover and the presence of millions of stereotypic crypts throughout the entire intestine.
Against the grain. In 2007, Nick Barker, a senior scientist in the Clevers lab, identified the Wnt target gene Lgr5 as a unique marker of adult stem cells in several epithelial organs, including the intestine, hair follicle, and stomach. In the intestine, the gene codes for a plasma membrane protein on crypt stem cells that enable the intestinal epithelium to self-renew, but can also give rise to adenomas of the gut. Upon making mice with adult stem cell populations tagged with a fluorescent Lgr5-binding marker, the lab helped to overturn assumptions that “stem cells are rare, impossible to find, quiescent, and divide asymmetrically.”
On to organoids. Once the lab could identify adult stem cells within the crypts of the gut, postdoc Toshiro Sato discovered that a single stem cell, in the presence of Matrigel and just three growth factors, could generate a miniature crypt structure—what is now called an organoid. “Toshi is very Japanese and doesn’t always talk much,” says Clevers. “One day I had asked him, while he was at the microscope, if the gut stem cells were growing, and he said, ‘Yes.’ Then I looked under the microscope and saw the beautiful structures and said, ‘Why didn’t you tell me?’ and he said, ‘You didn’t ask.’ For three months he had been growing them!” The lab has since also grown mini-pancreases, -livers, -stomachs, and many other mini-organs.
Tumor Organoids. Clevers showed that organoids can be grown from diseased patients’ samples, a technique that could be used in the future to screen drugs. The lab is also building biobanks of organoidsderived from tumor samples and adjacent normal tissue, which could be especially useful for monitoring responses to chemotherapies. “It’s a similar approach to getting a bacterium cultured to identify which antibiotic to take. The most basic goal is not to give a toxic chemotherapy to a patient who will not respond anyway,” says Clevers. “Tumor organoids grow slower than healthy organoids, which seems counterintuitive, but with cancer cells, often they try to divide and often things go wrong because they don’t have normal numbers of chromosomes and [have] lots of mutations. So, I am not yet convinced that this approach will work for every patient. Sometimes, the tumor organoids may just grow too slowly.”
Selective memory. “When I received the Breakthrough Prize in 2013, I invited everyone who has ever worked with me to Amsterdam, about 100 people, and the lab organized a symposium where many of the researchers gave an account of what they had done in the lab,” says Clevers. “In my experience, my lab has been a straight line from cloning TCF-1 to where we are now. But when you hear them talk it was ‘Hans told me to try this and stop this’ and ‘Half of our knockout mice were never published,’ and I realized that the lab is an endless list of failures,” Clevers recalls. “The one thing we did well is that we would start something and, as soon as it didn’t look very good, we would stop it and try something else. And the few times when we seemed to hit gold, I would regroup my entire lab. We just tried a lot of things, and the 10 percent of what worked, those are the things I remember.”
Greatest Hits
- Cloned the first T cell–specific transcription factor, TCF-1, and identified homologous genes in model organisms including the fruit fly, frog, and worm
- Found that transcriptional activation by the abundant β-catenin/TCF-4 [TCF7L2] complex drives cancer initiation in colon cells missing the tumor suppressor protein APC
- First to extend the role of Wnt signaling from developmental biology to adult stem cells by showing that the two Wnt pathway transcription factors, TCF-1 and TCF-4, are necessary for maintaining the stem cell compartments in the thymus and in the crypt structures of the small intestine, respectively
- Identified Lgr5 as an adult stem cell marker of many epithelial stem cells including those of the colon, small intestine, hair follicle, and stomach, and found that Lgr5-expressing crypt cells in the small intestine divide constantly and symmetrically, disproving the common belief that stem cell division is asymmetrical and uncommon
- Established a three-dimensional, stable model, the “organoid,” grown from adult stem cells, to study diseased patients’ tissues from the gut, stomach, liver, and prostate
Regenerative Medicine Comes of Age
“Anti-Aging Medicine” Sounds Vaguely Disreputable, So Serious Scientists Prefer to Speak of “Regenerative Medicine”
-
Induced pluripotent stem cells (iPSCs) and genome-editing techniques have facilitated manipulation of living organisms in innumerable ways at the cellular and genetic levels, respectively, and will underpin many aspects of regenerative medicine as it continues to evolve.
An attitudinal change is also occurring. Experts in regenerative medicine have increasingly begun to embrace the view that comprehensively repairing the damage of aging is a practical and feasible goal.
A notable proponent of this view is Aubrey de Grey, Ph.D., a biomedical gerontologist who has pioneered an regenerative medicine approach called Strategies for Engineered Negligible Senescence (SENS). He works to “develop, promote, and ensure widespread access to regenerative medicine solutions to the disabilities and diseases of aging” as CSO and co-founder of the SENS Research Foundation. He is also the editor-in-chief of Rejuvenation Research, published by Mary Ann Liebert.
Dr. de Grey points out that stem cell treatments for age-related conditions such as Parkinson’s are already in clinical trials, and immune therapies to remove molecular waste products in the extracellular space, such as amyloid in Alzheimer’s, have succeeded in such trials. Recently, there has been progress in animal models in removing toxic cells that the body is failing to kill. The most encouraging work is in cancer immunotherapy, which is rapidly advancing after decades in the doldrums.
Many damage-repair strategies are at an early stage of research. Although these strategies look promising, they are handicapped by a lack of funding. If that does not change soon, the scientific community is at risk of failing to capitalize on the relevant technological advances.
Regenerative medicine has moved beyond boutique applications. In degenerative disease, cells lose their function or suffer elimination because they harbor genetic defects. iPSC therapies have the potential to be curative, replacing the defective cells and eliminating symptoms in their entirety. One of the biggest hurdles to commercialization of iPSC therapies is manufacturing.
-
Building Stem Cell Factories
“Now that an infrastructure is being developed to make unlimited cells for the tools business, new opportunities are being created. These cells can be employed in a therapeutic context, and they can be used to understand the efficacy and safety of drugs,” asserts Chris Parker, executive vice president and CBO, Cellular Dynamics International (CDI). “CDI has the capability to make a lot of cells from a single iPSC line that represents one person (a capability termed scale-up) as well as the capability to do it in parallel for multiple individuals (a capability termed scale-out).”
Minimally manipulated adult stem cells have progressed relatively quickly to the clinic. In this scenario, cells are taken out of the body, expanded unchanged, then reintroduced. More preclinical rigor applies to potential iPSC therapy. In this case, hematopoietic blood cells are used to make stem cells, which are manufactured into the cell type of interest before reintroduction. Preclinical tests must demonstrate that iPSC-derived cells perform as intended, are safe, and possess little or no off-target activity.
For example, CDI developed a Parkinsonian model in which iPSC-derived dopaminergic neurons were introduced to primates. The model showed engraftment and enervation, and it appeared to be free of proliferative stem cells.
-
“You will see iPSCs first used in clinical trials as a surrogate to understand efficacy and safety,” notes Mr. Parker. “In an ongoing drug-repurposing trial with GlaxoSmithKline and Harvard University, iPSC-derived motor neurons will be produced from patients with amyotrophic lateral sclerosis and tested in parallel with the drug.” CDI has three cell-therapy programs in their commercialization pipeline focusing on macular degeneration, Parkinson’s disease, and postmyocardial infarction.
-
Keeping an Eye on Aging Eyes
The eye has multiple advantages over other organ systems for regenerative medicine. Advanced surgical methods can access the back of the eye, noninvasive imaging methods can follow the transplanted cells, good outcome parameters exist, and relatively few cells are needed.
These advantages have attracted many groups to tackle ocular disease, in particular age-related macular degeneration, the leading cause of blindness in the elderly in the United States. Most cases of age-related macular degeneration are thought to be due to the death or dysfunction of cells in the retinal pigment epithelium (RPE). RPE cells are crucial support cells for the rods, cones, and photoreceptors. When RPE cells stop working or die, the photoreceptors die and a vision deficit results.
A regenerated and restored RPE might prevent the irreversible loss of photoreceptors, possibly via the the transplantation of functionally polarized RPE monolayers derived from human embryonic stem cells. This approach is being explored by the California Project to Cure Blindness, a collaborative effort involving the University of Southern California (USC), the University of California, Santa Barbara (UCSB), the California Institute of Technology, City of Hope, and Regenerative Patch Technologies.
The project, which is funded by the California Institute of Regenerative Medicine (CIRM), started in 2010, and an IND was filed early 2015. Clinical trial recruitment has begun.
One of the project’s leaders is Dennis Clegg, Ph.D., Wilcox Family Chair in BioMedicine, UCSB. His laboratory developed the protocol to turn undifferentiated H9 embryonic stem cells into a homogenous population of RPE cells.
“These are not easy experiments,” remarks Dr. Clegg. “Figuring out the biology and how to make the cell of interest is a challenge that everyone in regenerative medicine faces. About 100,000 RPE cells will be grown as a sheet on a 3 × 5 mm biostable, synthetic scaffold, and then implanted in the patients to replace the cells that are dying or dysfunctional. The idea is to preserve the photoreceptors and to halt disease progression.”
Moving therapies such as this RPE treatment from concept to clinic is a huge team effort and requires various kinds of expertise. Besides benefitting from Dr. Clegg’s contribution, the RPE project incorporates the work of Mark Humayun, M.D., Ph.D., co-director of the USC Eye Institute and director of the USC Institute for Biomedical Therapeutics and recipient of the National Medal of Technology and Innovation, and David Hinton, Ph.D., a researcher at USC who has studied how actvated RPE cells can alter the local retinal microenvironment.
Read Full Post »













{kind=link}
{kind=link}