Organoid Development
Curator: Larry H Bernstein, MD, FCAP
Thyroid Organoids Made from Stem Cells Treat Thyroid-Deficient Mice
Darrell Kotton and his research team from Beth Deaconess Medical Center, in collaboration with researchers from the Boston University School of Medicine have devised a workable protocol for differentiating Human pluripotent stem cells into functional thyroid gland cells.
Every year, many people are diagnosed with an underactive thyroid and many others lose their thyroid as a result of thyroid cancer. Designing treatments that can help replace lost thyroid tissue would certainly be a welcome thing for these patients.
By working with mouse embryonic stem cells, Kotton and his colleagues showed that two growth factors, BMP4 and FGF2, and induce foregut endodermal cells to differentiate into thyroid cells. This simple signaling pathway not only efficiently generates thyroid tissue from endoderm, but this pathway turns out to be commonly used in species as diverse as frogs, mice and humans.
The BMP4/FGF2-treated foregut cells differentiated into small thyroid organics that Kotton and his team were able to transplant into thyroid-deficient mice. These transplantations restored normal thyroid function to these mice.

https://beyondthedish.files.wordpress.com/2015/10/fx1-6.jpg?w=652
{kind=link}
While mice cells are a fine model system for human diseases, they are not exactly the same. Can this procedure work with human cells? To answer that question, Kotton and his coworkers used human induced pluripotent stem cells (iPSCs) and subjected them to the same BMP4/FGF2 protocol after they had first differentiated the cells into endoderm. In addition, Kotton and his team made thyroid cells from iPSCs derived from cells taken from patients with a specific type of hypothyroidism (interactive thyroid). These patients lack a gene called NKX2-1, and suffer from congenital hypothyroidism.
The thyroid is responsible for your basal metabolic rate. Hypothyroidism or an interactive thyroid can cause patients to gain weight, feel tired constantly, have trouble concentrating, and have a slow heart rate. Hypothyroidism is usually treated with synthetic thyroid hormones that are taken orally. However, restoring a patient’s own thyroid tissue or even replacing defective thyroid tissue with repaired thyroid tissue would be a huge boon to thyroid patients.
This work has discovered the regulatory mechanisms that drive the establishment of the thyroid. It also provides a significant step toward cell-based regenerative therapy for hypothyroidism and the replacement of the thyroid after thyroid cancer treatments.
These results were published in the journal Cell Stem Cell, October 2015 DOI:10.1016/j.stem.2015.09.004.
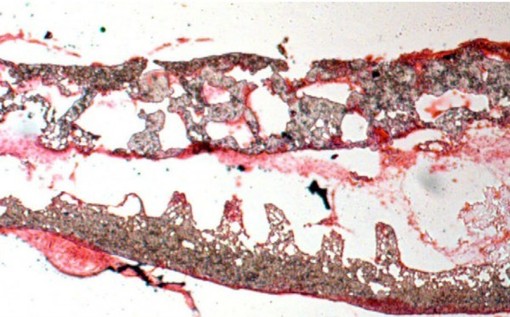
Artificial intestines created including nutrient-absorbing villi that might help humans with gut disorders
The idea of making replacement organs by growing cells on a scaffold is not new; other researchers have done so with bladders and blood vessels. But Hackam’s lab-grown intestine—described last week in Regenerative Medicine—comes closer to the shape and structure of a natural intestine than anything created before. In the past, gut lining has been grown on flat scaffolds or petri dishes, where it tended to curl into little balls with the food-absorbing surface trapped inside.
Hackam’s group overcame that with their scaffold, made from a material similar to surgical sutures that can be formed into any desired intestinal size and shape. Hackam’s scaffolds are tube-shaped like a real gut, with tiny projections on the inner surface to help the tissue grow into functional small intestine villi, tiny fingers of tissue that help absorb nutrients. “They can now make sheets of cells that can be clinically managed,” Clevers says. “Surgeons can handle these things and just stick them in.”
To grow the gut lining in the lab, the researchers painted the scaffold with a sticky substance containing collagen, dribbled it with a solution of small intestine stem cells, and then let it incubate for a week. They found that adding connective tissue cells, immune cells, and probiotics—bacteria that help maintain a healthy gut—helped stem cells mature and differentiate.

{kind=link}
Their gut lining regrew onto the scaffold and functioned normally to absorb water from the colon. Within weeks, the scaffolding dissolved and was replaced with normal connective tissue. “The scaffold was well tolerated and promoted healing by recruiting stem cells,” Hackam says. “[The dogs] had a perfectly normal lining after 8 weeks.”
Sourced through Scoop.it from: news.sciencemag.org
Pluripotent cells are promising tools in the arena of regenerative medicine. For many years, research efforts have been directed towards uncovering the underlying mechanisms that govern the pluripotent state and this involves identifying new pluripotency associated factors. Zinc finger protein 553 (Zfp553) has been hypothesized to be one such factor due to its predominant expression in inner cell mass of the mouse early embryo. In this study, we have identified Zfp553 as a regulator of pluripotency. Zfp553 knockdown down-regulates pluripotency markers and triggers differentiation in mouse embryonic stem cells (mESCs). Further investigation revealed that Zfp553 regulates pluripotency in mESCs via the transcriptional activation of Pou5f1 and Nanog. Microarray results revealed that depletion of Zfp553 down-regulates many pluripotency genes, as well as genes associated with metabolism related processes. ChIP-seq depicted the genomic binding sites of Zfp553 in mESCs and its binding motif. In addition, we found that depletion of Zfp553 could impair somatic cell reprogramming, evidenced by reduced reprogramming efficiency and cell viability. Together, our preliminary findings provide novel insights to a newly identified pluripotency factor Zfp553 and its role in pluripotency regulation.
Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs
Donald G. Phinney, Michelangelo Di Giuseppe, Joel Njah, Ernest Sala, Sruti Shiva, Claudette M. St Croix, Donna B. Stolz, Simon C. Watkins, Y. Peter Di, George D. Leikauf, Jay Kolls,… , & Luis A. Ortiz
- Nature Communications 07 Oct 2015; 6(8472) http://dx.doi.org:/10.1038/ncomms9472 http://www.nature.com/ncomms/2015/151007/ncomms9472/full/ncomms9472.html
Mesenchymal stem cells (MSCs) and macrophages are fundamental components of the stem cell niche and function coordinately to regulate haematopoietic stem cell self-renewal and mobilization. Recent studies indicate that mitophagy and healthy mitochondrial function are critical to the survival of stem cells, but how these processes are regulated in MSCs is unknown. Here we show that MSCs manage intracellular oxidative stress by targeting depolarized mitochondria to the plasma membrane via arrestin domain-containing protein 1-mediated microvesicles. The vesicles are then engulfed and re-utilized via a process involving fusion by macrophages, resulting in enhanced bioenergetics. Furthermore, we show that MSCs simultaneously shed micro RNA-containing exosomes that inhibit macrophage activation by suppressing Toll-like receptor signalling, thereby de-sensitizing macrophages to the ingested mitochondria. Collectively, these studies mechanistically link mitophagy and MSC survival with macrophage function, thereby providing a physiologically relevant context for the innate immunomodulatory activity of MSCs.
MSCs undergo mitophagy in response to oxidative stress
Human MSCs shed from their surface a diverse subpopulation of vesicles. To characterize these vesicles, we performed electron microscopy on those recovered from MSC-conditioned medium by differential ultracentrifugation (100,000 g for 18 h). This analysis demonstrated the presence of 50–100-nm vesicles that are morphologically consistent with exosomes (Fig. 1a, left). Flotation of the 100,000 g pellets on sucrose gradients followed by western blot and fluorescent activated cell sorting (FACS) further demonstrated that these vesicles expressed the exosomal markers’ milk fat globule factor 8 (Mfge8) and the tetraspanins CD9 and CD63, respectively (Supplementary Fig. 1A). Centrifugation of conditioned medium at low speeds (10,000 g) revealed the presence of larger vesicles (>100 nm) that contain subcellular mitochondrial structures including outer and inner membranes and cristae, and expressed the mitochondria-specific protein ATP synthase as evidenced by immuno-gold labelling (Fig. 1a, centre and Supplementary Fig. 1B). MSCs also release larger multivesicular bodies containing lysosome-like vesicles and entire mitochondria, suggesting that these organelles were selected for mitophagy by targeting to autophagosomes (Fig. 1a, right).

(a) Left panel, electron microscopy of vesicles isolated from sucrose densities of 1.11 and 1.14 g ml−1 and purified using differential ultracentrifugation (100,000g per 18 h) reveal a typical exosome morphology. Middle panel, MVs over 100 nm in size recovered from human MSC-conditioned medium following low-speed (10,000 g per 1 h) centrifugation contain structures conforming to the morphology of mitochondria. Right panel, MVs contain closely packed-vesicles and entire mitochondria (multivesicular body (MVB)) representing autophagosomes. (b) Left panel, flow cytometric analysis of MitoSOX Red-stained human MSCs expanded in 5 or 21% oxygen for 7 days. Right panel, quantification of the flow cytometric data. Plotted values (mean±s.e.m.) represent four replicates for each sample using three distinct replicate cultures from each experimental group. (c) Mitochondrial membrane potential of human MSCs from bdetermined using JC-1 staining. Expansion in 21% oxygen results in partial depolarization of mitochondria as evidenced by accumulation of JC-1 monomers (*P<0.005, Student’s-t test versus MSCs in 5%oxygen). (d) Western blot analysis (Supplementary Fig. 1) of cytoplasmic or mitochondrial extracts prepared from human MSCs expanded in 21% oxygen for the indicated passage numbers (P1 or P4) reveals Parkin mitochondrial translocation and Pink1 kinase activation in human MSCs but not human dermal fibroblasts. Data are representative of a single experiment repeated five times.
To determine whether culture expansion of MSCs impairs mitochondrial function, we expanded cells under physiological oxygen levels (5% O2) or standard culture conditions (21% O2) and quantified mtROS levels and mitochondrial membrane potential by staining with MitoSOX Red and JC-1, respectively37. FACS analysis of stained cells confirmed that exposure to 21% oxygen resulted in a significant increase in mtROS (Fig. 1b) and a concomitant decrease in mitochondrial membrane potential as demonstrated by the accumulation of JC-1 monomers (Fig. 1c). Moreover, western blot analysis revealed that prolonged exposure to 21% oxygen activated the Pink1/Parkin-mediated pathway of mitophagy in MSCs but not in human fibroblasts cultured under identical conditions (Fig. 1d). Herein, increased mitochondrial expression of Parkin and Pink1 kinase was accompanied by Pink1 kinase activation as evidenced by the presence of lower molecular weight moieties of the protein and reduced Miro levels in mitochondrial extracts from P4 versus P1 MSCs (Fig. 1c and Supplementary Fig. 1C). Importantly, Pink1 targets Miro for degradation, thereby severing the connection of the mitochondria to the cytoskeleton and facilitating its incorporation into the phagosome38. Consistent with these results, western blot analysis also revealed that mitochondria-containing MVs expressed microtubule-associated protein 1 light chain 3 (LC3) and autophagy-related protein 12, which are highly enriched in MVs as compared with whole-cell extracts (Supplementary Fig. 1D). Therefore, these MVs are characteristic of autophagosomes39.
MSCs package mitochondria in MVs for cellular transfer
To examine mitophagy in MSCs in more detail, MSCs were infected with baculoviruses encoding green fluorescent protein (GFP) fused to the E1alpha pyruvate dehydrogenase leader peptide, which drives transport to the mitochondria, and LC3 fused to a red fluorescent protein (RFP) to allow tracking to the phagophore39, 40. Fluorescent microscopy confirmed that the GFP-labelled mitochondrial network is in close proximity to RFP-LC3-labelled phagosomes (Fig. 2a). Live cell imaging further revealed that mitochondria are loaded in the cytoplasm into LC3-containing vesicles, which migrate towards the cell periphery and are incorporated into outward budding blebs in the plasma membrane (Fig. 2b–d and Supplementary Movie 1). Western blot analysis further revealed that RFP-LC3-MVs also expressed the endosomal sorting complex required for transport (ESCRT)-associated proteins’ tumour suppressor gene 101 (TSG101) and arrestin domain-containing protein 1 (ARRDC1)11, 41 (Supplementary Fig. 1D). Collectively, these results indicate that MSCs employ the release of ARMMs to extrude mitochondria at their cell surface. Moreover, MSCs exhibited marked increases in apoptosis when treated with Bafilomycin A1 or low concentrations (3–5 μM) of chloroquine, which block the mitophagy flux, indicating that this process is critical for MSC survival (Supplementary Movie 2).

(a–d) Differential interference contrast (DIC) fluorescence overlay of live human MSCs expressing fluorescent proteins that target mitochondria (green) and phagosomes (red) shows mitochondria being loaded into phagosomes (arrows), which are then shuttled to the plasma membrane for extrusion (also seeSupplementary Movie 1). (e–h) Inset shows a representative macrophage interacting with a human MSC. This interaction is shown as a time sequence (5 min intervals) in the lower images and in Supplementary Movie 3. The inset demarcates the area in the human MSC plasma membrane where the membrane blebs outwards and accumulates vesicles. Macrophages nibble the surface of human MSCs and uptake mitochondrial laden phagosomes from blebs budding (arrows) from the plasma membrane of the human MSCs. Scale bars, 10 μ.
Next, we co-cultured GFP-labelled human MSCs from above with primary human or mouse macrophages. Live cell imaging revealed that macrophages nibble the plasma membrane of MSCs, establishing cell contact at areas where membrane blebs are enriched in RFP-labelled vesicles, which are subsequently stripped by the macrophage (Fig. 2e–h and Supplementary Movie 3). This activity was also observed between mouse macrophages and primary human MSCs (Fig. 3a and Supplementary Movies 4 and 5) but was not evident when macrophages were co-cultured with mouse or human fibroblasts (Supplementary Fig. 2). In a subsequent experiment, we co-cultured the macrophage cell line RAW 264.7 with human MSCs containing RFP-labelled mitochondria (10:1 ratio) for 4 h and recovered macrophages using FACS after staining with antibodies that recognize macrophage epitopes (that is, F4/80) not expressed by MSCs. Sorted macrophages were cultured for up to 2 weeks in RPMI media, which do not support MSC expansion and survival. Fluorescent microscopy of these macrophages revealed clear evidence of cell-associated RFP derived from human MSCs (Fig. 3b). To confirm these findings, we demonstrated using PCR amplification that these macrophages expressed the mitochondrial specific transcript human cytochrome c oxidase I (MT-COX I), which was confirmed on the basis of the restriction fragment pattern obtained after digestion of the PCR product with Bfa1 (Fig. 3b). This PCR product was not detected in mouse macrophages because of limited sequence homology between the two genes30 but was detected in human MSC-derived MVs as expected (Fig. 3c and Supplementary Fig. 3A). Lastly, we co-cultured Cy5-labelled human MSCs with macrophages that were pre-incubated with or without dextran sulfate (100 μg ml−1), a nonspecific inhibitor of phagocytosis. Live cell imaging showed phagocytosis of MVs by macrophages over a period of 18 min, and confocal microscopy confirmed that the engulfed Cy5-labelled vesicles resided within the cell body of the macrophage (Supplementary Movie 6). However, MV uptake was blocked in macrophages pre-treated with dextran sulfate as evidenced by the accumulation of Cy5-labelled MVs on the macrophage surface (Supplementary Movie 7).

(a) Top panel is DIC fluorescent overlay at time 0 of primary human MSCs infected with Organelle Lights to label mitochondria (green) and co-cultured with mouse (RAW 264.7) macrophages. Lower panels, time sequence at 45 min intervals showing transfer of green-labelled mitochondria from the a MSC to a macrophage (red arrow, see Supplementary Movie 4 for transfer of mitochondria in filamentous form, andSupplementary Movie 5 in which GFP signal is compensated to allow the tracking of the transferred mitochondria into macrophages). (b) Left panel, photomicrograph of FACS-sorted mouse macrophages that were co-cultured with mitochondria-labelled (RFP) human MSCs clearly show retention of RFP label. Right panel, electrophoretic pattern of human COX I PCR product treated with or without Bfa1 after amplification from the indicated cell sources. (c) MSC-derived exosomes and MVs express the Bfa1-sensitive 228-bp COX I mtDNA PCR product detected in human MSCs (b). (d) Left panel, electrophoretic pattern of Bfa1-digested human COX1 PCR product amplified from mouse lung DNA isolated 14 days after the intravenous administration of human MSCs, human MSC-derived MVs or exosomes. Right panel, human GAPDH and human COX1 relative expression levels quantified by RT–PCR in mouse lung (3–28 days) after a single (intratracheal (IT) or intravenous (IV)) injection of human MSCs, human MSC-derived exosomes or human fibroblasts. *P<0.001, #P<0.001 by ANOVA compared with untreated mouse lung. Plotted values (mean±s.e.m.) are from experiments repeated four times. Scale bars, 20 μ.
To track the in vivo transfer of mitochondria, we systemically administered RFP-labelled human MSCs into C57BL/6 mice expressing a GFP reporter under control of the endothelial specific Tie2 promoter. At 24 h post injection, GFP-labelled endothelial cells, epithelial cells and macrophages that contained RFP-labelled mitochondria were visible (Supplementary Fig. 3B). Bfa1 digestion of mouse lung DNA following intravenous administration of human MSCs, exosomes or MVs yielded a pattern of restriction similar to those observed in RAW 264.7 macrophages (Fig. 3d). To follow the fate of viable human MSCs in the mouse lung, we measured the abundance of human-specific GAPDH transcripts via reverse transcriptase–PCR (RT–PCR)42. Human GAPDH mRNA was not detected in the lung tissue of untreated mice but was detected at 3 days post injection of human MSCs or human fibroblasts (Fig. 3d). However, expression rapidly declined and was no longer evident by 14 or 28 days post transplant, consistent with the clearance rate of cells from lung tissue. Expression of human COXI mRNA in mouse lung mirrored that of human GAPDH following injection of human fibroblasts and was detected at 3 days but not 14 or 28 days post transplant. In contrast, human COXI transcripts were detected up to 28 days post injection of human MSCs, indicating that mouse lung tissue retained mtDNA long after the disappearance of viable human MSCs (Fig. 3d). Thus, MSC-derived vesicles constitute an effective mechanism to transfer mtDNA into the mouse lung.
MSC extracellular vesicles enhance macrophage energetics
To study the effect of MVs on macrophage bioenergetics, we analysed oxygen consumption rates (OCRs) using the SeaHorse technology. Human macrophages exhibit higher basal OCR than human MSCs or human fibroblasts (Fig. 4a). Co-culture of macrophages with human MSCs (Mac+hMSC) or MSC-derived exosomes (Mac+Exo) but not human fibroblasts (Mac+Fibro) significantly (analysis of variance (ANOVA) followed by Student–Neuman Keuls (SNK) post-hocpairwise comparisons) increased their OCR, suggesting that MSCs or MSC-derived exosomes alter macrophage bioenergetics (Fig. 4a). Next, we repeated these measurements after treatment of cells with oligomycin A, an inhibitor of ATP synthase, which is required for the oxidative phosphorylation of ADP to ATP. These conditions differentiate ATP-linked respiration from the proton leak. Macrophages exhibited a higher level of proton leak as compared with human MSCs and fibroblasts, and proton leak was significantly (ANOVA followed by SNK post hoc pairwise comparisons) reduced following co-culture with human Mac+Exo but not Mac+Fibro (Fig. 4a). Co-culture with human MSCs (Mac+hMSC) also significantly (ANOVA followed by SNK post hocpairwise comparisons) reduced proton leak in macrophages. We also repeated the OCR measurements following treatment of cells with the uncoupling agent carbonyl cyanide 4-(trifluoromethoxy) phenylhydraone (FCCP) to determine how cells respond to an increase in ATP demand. All three cell types responded to FCCP treatment with increased OCR, and the magnitude of the response was greater in macrophages as compared with human MSCs and fibroblasts. Moreover, OCR was significantly increased in FCCP-treated macrophages following co-culture with human MSCs (Mac+hMSC) or human Mac+Exo but not Mac+Fibro (Fig. 4a).

(a) Mitochondrial respiration of human macrophages, human MSCs or human fibroblasts was measured as OCR using the XF technology. Macrophages were co-cultured with or without human MSCs or fibroblasts (1:10 ratio) or treated with human MSC-derived exosomes (40 μg per protein) in the presence or absence of Oligomycin A and FCCP to differentiate ATP-linked respiration from the proton leak. Plotted data (mean±s.e.m.) were performed using six replicates per sample and repeated three times. (b) Pseudocoloured photomicrographs (0–240 min) of MitoSOX Red-stained macrophages that were non-stimulated (upper panel), or treated with silica (20 μg cm−2, lower panel) or silica plus human MSC-derived exosomes (added 10 min after silica, middle panel). Scale bars, 50 μ. (c) Time course of MitoSOX Red emission by human macrophages treated as in b. Figure is representative of five exposures (nine stages positions per test and 6 cells per stage). (d) OCR as in a of silica-exposed macrophages treated with or without human MSCs, human MSC-derived exosomes or human fibroblasts. Plotted values (mean±s.e.m.) are from experiments repeated three times, *P<0.05 as compared to control, #P<0.05 as compared to silica treated macrophages, as determined by Student’s t-test.
To examine the effect of MSC or exosomes on macrophage bioenergetics under conditions of altered homeostasis, we exposed macrophages to silica particles. Silica exposure results in a burst of mtROS production as evidenced by changes in MitoSOX Red fluorescence intensity; however, this effect is largely mitigated in macrophages incubated with human MSC-derived exosomes (Fig. 4b,c). Silica exposure also decreased macrophage OCR, but this decrease was reversed by co-culture with human MSCs or human MSC-derived exosomes but not with human fibroblasts (Fig. 4d). The fact that transfer of partially depolarized mitochondria from MSCs to macrophages enhances that macrophage bioenergetics appears paradoxical. However, loss of mitochondrial membrane potential as a result of MSC expansion is not absolute as mitochondria exhibit residual membrane potential as evidenced by the concentration of JC-1 aggregates (Fig. 1c). This indicates that the mitochondrial membrane is not collapsed and the mitochondria are still capable of undergoing fusion. To determine whether these mitochondria are recycled in macrophages by fusion, we co-cultured human MSCs with macrophages after labelling cells with two different MitoTracker dyes (Red and Green)43. Live cell imaging clearly demonstrated the transfer and subsequent fusion (yellow colour in merged images) of RFP-labelled, human MSC-derived mitochondria with GFP-labelled mitochondria within human macrophages (Fig. 5). These data indicate that under oxidative stress MSCs outsource mitophagy to macrophages to unload partially depolarized mitochondria, which are recycled via fusion by macrophages thereby enhancing their bioenergetics.

Human MSCs and human macrophages (1 × 105) were infected separately with Organelle Lights to label human MSC mitochondria (red) and macrophage mitochondria (green). Twenty-four hours following infection, macrophages were harvested and co-incubated with the human MSCs for 2 h. Images were collected using an inverted Nikon TiE fluorescent microscope equipped with a × 60 oil immersion optic and NIS Elements Software. Organelle Lights were excited using a Lumencor diode-pumped light engine and detected using an ORCA-Flash4.0 sCMOS camera. (a,b) DIC images of two separate fields within the same dish. (c) A zoomed image of the outlined section within b (scale bars, 20 μ). The fluorescence-based images for each field appear in the panels below the DIC images, with d–f showing macrophage mitochondria (green); g–i showing human MSC mitochondria (red); and j–l showing the overlay with yellow indicative of colocalization of human MSC and macrophage mitochondria. Not every macrophage was shown to take up human MSC mitochondria (a,d,g, j).
MSC-derived exosomes are enriched in microRNAs
Exosomes transfer RNAs between cells8. We hypothesized that this process may be exploited by MSCs to tolerize macrophages against mitochondrial transfer. To explore this possibility, we analysed the RNA content of human MSC-derived exosomes. Using microRNA microarray analysis, we identified 156 (45 increased; 111 decreased) microRNAs that differed (log2>1.0,P<0.05 (ANOVA followed by Holm–Sidak post hoc pairwise comparisons) in abundance between exosomes compared with their parent MSCs. The 10 microRNAs that exhibited the greatest increase included miR451a (316-fold), miR1202 (45-fold), miR630 (40-fold) and miR638 (28-fold), while microRNAs that exhibited the greatest decrease in exosomes and were enriched in MSCs included miR125b (148-fold) and miR21 (91-fold; Fig. 6a,b). This pattern of microRNA expression was conserved in MSC-derived exosomes obtained from five human donors (Fig. 6c,d).

(a) Heatmap illustrating the 10 microRNAs most highly enriched in human MSCs versus their corresponding exosomes. Every row represents a microRNA and every column a cell or exosome, and yellow and purple represent increased or decreased expression, respectively. (b) Plotted values represent the means log 2 fold enrichment of exosomal versus human MSC microRNAs (n=5 microarrays of different MSCs cell lines; P<0.05, ANOVA followed by Holm–Sidak post hoc pairwise comparisons). (c) Data in b show distribution of differentially expressed microRNAs between samples based on the –log base 10 significant P value (<0.05) and with a relative fold change of >2 (in log base 2). Green and red squares represent increasingly and decreasingly expressed microRNAs, respectively, in exosomes versus human MSCs. (d) Computational analysis of human MSCs and exosomes from five donors demonstrates that microRNAs isolated in exosomes cluster among different donors.
MSC-derived exosomes inhibit TLR signalling in macrophages
Mitochondrial uptake can induce inflammation via activation of pattern recognition receptors34. Therefore, given the presence of mtDNA and microRNAs in MSC-derived MVs and exosomes, respectively, we hypothesized that exposure to these vesicles would tolerize macrophages to mitochondrial transfer by inducing changes in TLR expression. Subsequently, we profiled the expression of 84 TLR-associated transcripts in mouse macrophages. We contrasted these results with those observed in macrophages that were co-cultured with mouse or human MSCs, human MSC-derived exosomes or silica particles, which when phagocytized induce macrophage activation44. Co-culture of macrophages with MSC-derived exosomes induced nuclear translocation of the transcription factor NF-κB (Fig. 7a) resulting in significant changes (>2.5-fold increase or decrease) in expression of 50 of the 84 TLR-associated transcripts (Fig. 7b). For example, compared with silica-exposed macrophages those treated with exosomes exhibited significant (>2.5-fold) increases in transcripts associated with cytokine signalling including interleukin (IL)-1β, prostaglandin endoperoxide synthase 2 (PTGS2, aka COX2), granulocyte colony-stimulating factor 3 (CSF3), IL-10, chemokine (C–C motif) ligand 2 (CCL2, aka MCP-1), NF-κB-chemokine (C–X–C motif) ligand 10 (CXCL10), tumour necrosis factor (TNF) and reticuloendotheliosis oncogene (Rel; Fig. 7b). In contrast, transcripts encoding proteins involved in MyD88-dependent signalling (MyD88, TLR 1,4,5,7,8 and 9, IRAK1 and TRAF6), TRIF-dependent signalling (TLR adaptor molecule 1 (TICAM1) and TICAM2) and TLR-related signalling (CD80, CD86, IL-2, IL-12, Interferon gamma, PGLYRP1 and CSF2) were downregulated.

(a) Upper panels, confocal microscopy showing intracellular localization of Cy5-labelled exosomes within macrophages 18 min post administration. Lower panel, nuclear localization of NF-κB in macrophages 2 h post administration of exosomes. Scale bars, 15 μ. (b) Partial heatmap illustrating mRNA levels of 84 TLR-associated transcripts in macrophages at 8 h post treatment with silica (20 mg cm−2), human or mouse MSCs (1:10 ratio) or human MSC-derived exosomes (40 μg protein). Transcript order is highest (top) to lowest (bottom), and each row represents a gene and each column a specific treatment. Red and green illustrates increased or decreased gene expression, respectively. Experiments were repeated four times. (c) Effect of exosome treatment on PGE2, TNF and IL-10 secretion in macrophages from the indicated mouse strains. Plotted data (mean±s.e.m.) were from experiments repeated five times. *P<0.05 compared with C57BL/6J or BALB/CJ macrophages as determined by ANOVA). (d) Upper panel, western blot illustrating the time-dependent effect of silica or human MSC-derived exosomes on expression of TLR7 in macrophages. Lower panel, fold change in expression of the indicated transcripts in macrophages RT–PCR demonstrating the negative regulation of exosomes on macrophage expression of TLR genes. Plotted data (mean±s.e.m.) are from experiments repeated four times. *P<0.05 compared with baseline.#P<0.05 compared with native exosomes by Student’ t-test. †P<0.05 compared with the effect of native exosomes and indomethacin (Indo) treated by Student’s t-test.
MSCs secrete PGE2 that acts on prostaglandin receptors of LPS-stimulated macrophages to enhance their production of the anti-inflammatory cytokine IL-10 (ref. 45). However, this effect of MSCs was abrogated in macrophages from TLR4, MyD88, TNFR1 or COX2-deficient mice45. Consistent with these results, exosome treatment of non-stimulated macrophages augmented secretion of PGE2, TNF, IL-10 and IL-1-receptor antagonist (Fig. 7c), which may reprogramme macrophages3. These responses recapitulate those observed when macrophages are exposed to intact human or mouse MSCs, except that IL-6, CSF2 and IL-1 receptor 1 were increased more following exposure to mouse MSCs (Fig. 7b).
Subsequently, we treated TLR-signalling-deficient macrophages (TLR4−/−, TLR9−/−, MyD88−/−) or scavenger receptor-deficient macrophages (MARCO−/−) with MSC-derived exosomes. As shown inFig. 7c, PGE2 production was similar following exosome treatment in all signalling-deficient macrophages as compared with wild-type cells from strain-matched C57BL/6J or BALB/CJ mice. In contrast, secretion of TNF and IL-10 was significantly (ANOVA followed by SNK post hoc pairwise comparisons) reduced in TLR4−/− and MYD88−/− macrophages as compared with wild-type cells following exosome treatment, and IL-10 secretion was also significantly reduced in macrophages from TLR9−/− mice (Fig. 7c). These data confirm the importance of TLRs and in particular MyD88-dependent pathways in mediating exosome-induced effects on macrophage function. Lastly, we showed that pre-incubation with dextran sulfate significantly (ANOVA followed by SNK post hocpairwise comparisons) reduced the release of PGE2, TNF and IL-10 by exosome-treated macrophages, confirming the need for phagocytosis of MSC-derived vesicles in this process (Supplementary Fig. 4A).
To examine the role of microRNAs in macrophage tolerization, we treated RAW 264.7 cells, which use TLRs to recruit autophagy proteins in phagosomes to degrade its cargo46, with exosomes derived from human MSCs transfected with or without an short-hairpin RNA (shRNA) designed to inhibit DICER expression in the presence or absence of indomethacin, a cyclooxygenase inhibitor (Supplementary Fig. 4B). Treatment of naive RAW 264.7 macrophages with native exosomes enhanced TNF and reduced TLRs and MyD88 mRNA expression over 24 h (Fig. 7c), while treatment of silica-exposed macrophages with exosomes ameliorated TLR7 induction following silica exposure (Fig. 7c). Pre-incubation of RAW 264.7 macrophages with indomethacin before treatment with native exosomes, or treatment with exosomes from DICER knockout MSCs significantly (ANOVA followed by SNK post hoc pairwise comparisons) reduced the observed effects on TLR mRNA expression (Fig. 7c) and reduced secretion of proteins such as TNF, MIP, MCP1, KC and IP-10 associated with macrophage activation (Supplementary Fig. 4B,C). The inhibitory effects of indomethacin were restricted to TLR4 and MyD88 mRNA, while the effects of DICER-deficient exosomes were of greater magnitude and also involved negative regulation of TLR 7 and 9 (Fig. 7c). Concomitant treatment with indomethacin and exosomes from DICER-deficient exosomes demonstrated additive effects (Fig. 7c).
Importantly, miR-451 is one of the most abundantly expressed microRNAs in MSC-derived exosomes, but its maturation occurs independent of DICER47. Therefore, its expression is not altered in exosomes from DICER knockdown MSCs. MiR-451 negatively regulates cytokine production in dendritic cells infected with influenza virus48. Consistent with these results, transfection of RAW 264.7 macrophages with a miR-451 mimic significantly (Student’s t-test) decreased TNF mRNA expression in non-stimulated macrophages, and inhibited mRNA expression and protein release in silica-exposed macrophages (Supplementary Fig. 4D). In contrast, treatment of cells with a miR-451 antagomir yielded the opposite result. These data confirm a role of exosome-derived microRNAs in regulating cytokine expression in macrophages.
MSC exosomes attenuate monocyte activation and silicosis
Circulating MVs enter the bone marrow and reprogramme cells to express proteins of the tissue of vesicle origin49. Ly6Chi monocytes are recruited from the bone marrow into the lung in response to injury and play an important role in the pathogenesis of lung fibrosis50, 51. Therefore, we investigated whether MSC or their exosomes are capable of altering the lung recruitment and cytokine production of Ly6Chi monocytes in mice following silica exposure. As shown in Fig. 8a, FACS identified a limited number of Ly6Chi monocytes in the normal mouse lung, which was significantly (ANOVA followed by SNK post hoc pairwise comparisons) increased by 72 h post exposure to silica. Moreover, high expression of CCR2 and release of inflammatory (TNF, CCL2 and CXCL1) and fibrotic (transforming growth factor β (TGFβ) and IL-10) mediators indicate that these monocytes are activated (Fig. 8b). In contrast, intravenous administration of human MSCs (500,000 cells) or freshly isolated human MSC-derived exosomes (~3 × 1011 exosomes containing 40 μg protein) at 24 h post-silica exposure (0.2 g·kg−1) significantly reduced the extent of Ly6Chimonocyte infiltration into the lung and secreted levels of inflammatory mediators (Fig. 8a,b).

(a) Upper panel, absolute number of F4/80/CD11b- and Ly6C/CCR2-expressing cells in lung tissue of mice 72 h post administration of saline (50 μl), silica (0.2 g kg−1) or silica plus human MSC-derived exosomes (~3 × 1011 exosomes containing 40 μg protein). *P<0.05 compared with saline by t-test). Lower panel, representative histograms of flow cytometric data analysed in a showing the phenotype and frequency of cells recovered from lung tissue after enzymatic digestion. (b) Mulitplex ELISA of inflammatory (TNF, MCP1 and KC) and fibrotic (TGFβ and IL-10) mediators secreted by cultured F4/80/CD11b/ and Ly6C/CCR2 cells from a. Plotted values (mean±s.e.m.) are from experiments usingN=5 animals per group and repeated three times. *P<0.05 compared with saline, #P<0.001 compared with silica-treated monocytes by ANOVA.
Silica induces inflammation and collagen deposition in peri-bronchiolar, silicotic nodule and peri-vascular regions of the lung (Fig. 9a). These lesions are associated with enhanced numbers of inflammatory cells (although the percentage of macrophages decreases, there is an increase in neutrophils and lymphocytes) in bronchoalveolar lavage fluid (BALF, Fig. 9b), significant deposition of lung collagen as measured by hydroxyproline (Fig. 9c) and enhanced expression of pro-inflammatory cytokines (TNF and IL-6) and fibrotic mediators (IL-10 and α(I) collagen) by 14 and 28 days after silica exposure (Fig. 9d). Intravenous administration of human MSCs or exosomes 3 days after silica exposure reduced the size of the silicotic nodules (Fig. 9a), the total number of white blood cells in BALF (Fig. 9b) and expression of inflammatory and pro-fibrotic genes in the lung (Fig. 9d). Administration of exosomes significantly (ANOVA followed by SNK post hocpairwise comparisons) reduced the accumulation of neutrophils and lymphocytes in BALF, while MSCs only reduced the accumulation of neutrophils and induced a slight (<1%) increase in eosinophil count (Fig. 9b). In contrast, the intravenous administration of human fibroblasts significantly exacerbated the inflammatory and fibrotic responses to silica (Fig. 9a–c). Exosomes, but not MSC or fibroblast administration, reduced the accumulation of hydroxyproline in lung tissue 28 days after silica (Fig. 9c).

(a) Photomicrographs of lung sections stained with haematoxylin and eosine from mice 28 days after intratracheal administration of silica (0.2 g kg−1) alone or followed 3 days later with an intravenous injection of human MSCs, human MSC-derived exosomes (~3 × 1011 exosomes containing 40 μg protein) or human fibroblasts (scale bars, 500 μ). (b) Upper panel, photomicrographs of Diff-Quick-stained cytospins of BAL from mice in a. Lower panel, differential cell counts showing counts of total cells (left) and percentage of macrophages, lymphocytes, neutrophils and eosinophils (right panels). *P<0.05 compared with control, †P<0.05 compared with fibroblasts treated mice by Student’s t-test. (c) Hydroxyproline content of lung tissue from animals treated as in a. *P<0.001 compared with saline by Student’s t-test.†P<0.05 compared with silica, human MSC or fibroblast by ANOVA. (d) Quantification of mouse TNF, IL-6, IL-10 and Col1α1 levels in lung tissue from mice in a at 14 and 28 d post treatment. Plotted values (mean±s.e.m.) are representative of experiments using 15 animals per group and repeated three times.*P<0.001 compared with saline by Student’s t-test, †P<0.05 compared with silica by ANOVA.
Leave a Reply