Developing Deep Learning Models (DL) for the Instant Prediction of Patients with Epilepsy
Reporter: Srinivas Sriram, Research Assistant I
Research Team: Srinivas Sriram, Abhisar Anand
2021 LPBI Summer Intern in Data Science and Website Construction
This article reports on a research study conducted from January 2021 to May 2021.
This Research was completed before the 2021 LPBI Summer Internship that began on 6/15/2021.
The main criterion of this study was to utilize the dataset (shown above) to develop a DL network that could accurately predict new seizures based on incoming data. To begin the study, our research group did some exploratory data analysis on the dataset and we recognized the key defining pattern of the data that allowed for the development of the DL model. This pattern of the data can be represented in the graph above, where the lines representing seizure data had major spikes in extreme hertz values, while the lines representing normal patient data remained stable without any spikes. We utilized this pattern as a baseline for our model.
Conclusions and Future Improvements:
Through our system, we were able to create a prototype solution that would predict when seizures happened in a potential patient using an accurate LSTM network and a reliable hardware system. This research can be implemented in hospitals with patients suffering from epilepsy in order to help them as soon as they experience a seizure to prevent damage. However, future improvements need to be made to this solution to allow it to be even more viable in the Healthcare Industry, which is listed below.
- Needs to be implemented on a more reliable EEG headset (covers all neurons of the brain, less prone to electric disruptions shown in the prototype).
- Needs to be tested on live patients to deem whether the solution is viable and provides a potential solution to the problem.
- The network can always be fine-tuned to maximize performance.
- A better alert system can be implemented to provide as much help as possible.
These improvements, when implemented, can help provide a real solution to one of the most common diseases faced in the world.
Background Information:
Epilepsy is described as a brain disorder diagnostic category for multiple occurrences of seizures that happen within recurrent and/or a brief timespan. According to the World Health Organization, seizure disorders, including epilepsy, are among the most common neurological diseases. Those who suffer seizures have a 3 times higher risk of premature death. Epilepsy is often treatable, especially when physicians can provide necessary treatment quickly. When untreated, however, seizures can cause physical, psychological, and emotional, including isolation from others. Quick diagnosis and treatment prevent suffering and save lives. The importance of a quick diagnosis of epilepsy has led to our research team developing Deep Learning (DL) algorithms for the sole purpose of detecting epileptic seizures as soon as they occur.
Throughout the years, one common means of detecting Epilepsy has emerged in the form of an electroencephalogram (EEG). EEGs can detect and compile “normal” and “abnormal “brain wave activity” and “indicate brain activity or inactivity that correlates with physical, emotional, and intellectual activities”. EEG waves are classified mainly by brain wave frequencies (EEG, 2020). The most commonly studied are delta, theta, alpha, sigma, and beta waves. Alpha waves, 8 to 12 hertz, are the key wave that occurs in normal awake people. They are the defining factor for the everyday function of the adult brain. Beta waves, 13 to 30 hertz, are the most common type of wave in both children and adults. They are found in the frontal and central areas of the brain and occur at a certain frequency which, if slow, is likely to cause dysfunction. Theta waves, 4 to 7 hertz, are also found in the front of the brain, but they slowly move backward as drowsiness increases and the brain enters the early stages of sleep. Theta waves are known as active during focal seizures. Delta waves, 0.5 to 4 hertz, are found in the frontal areas of the brain during deep sleep. Sigma waves, 12-16 hertz, are very slow frequency waves that occur during sleep. EEG detection of electrical brain wave frequencies can be used to detect and diagnose seizures based on their deviation from usual brain wave patterns.
In this particular research project, our research group hoped to develop a DL algorithm that when implemented on a live, portable EEG brain wave capturing device, could accurately predict when a particular patient was suffering from Epilepsy as soon as it occurred. This would be accomplished by creating a network that could detect when the brain frequencies deviated from the normal frequency ranges.
The Study:
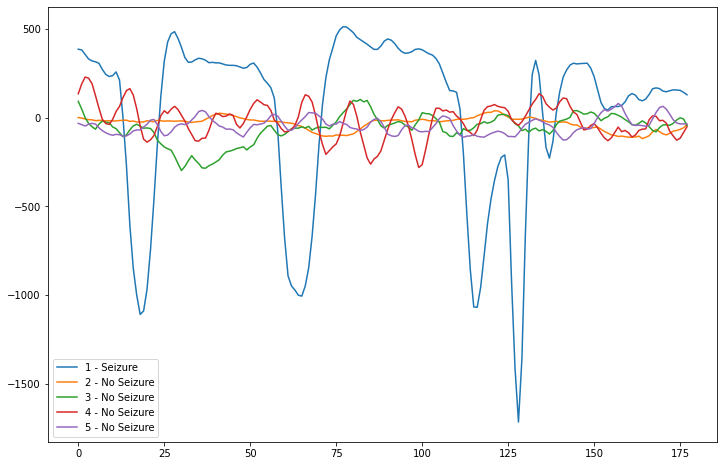
Line Graph representing EEG Brain Waves from a Seizure versus EEG Brain Waves from a normal individual.
Source Dataset: https://archive.ics.uci.edu/ml/datasets/Epileptic+Seizure+Recognition
To expand more on the dataset, it is an EEG data set compiled by Qiuyi Wu and Ernest Fokoue (2021) from the work of medical researchers R.Andrzejak, M.D. et al. (2001) which had been made public domain through the UCI Machine Learning Repository We also confirmed fair use permission with UCI. The dataset had been gathered by Andrzejak during examinations of 500 patients with a chronic seizure disorder. R.G.Andrzejak, et al. (2001) recorded each entry in the EEG dataset used for this project within 23.6 seconds in a time-series data structure. Each row in the dataset represented a patient recorded. The continuous variables in the dataset were single EEG data points at that specific point in time during the measuring period. At the end of the dataset, was a y-variable that indicated whether or not the patient had a seizure during the period the data was recorded. The continuous variables, or the EEG data, for each patient, varied widely based on whether the patient was experiencing a seizure at that time. The Wu & Fokoue Dataset (2021) consists of one file of 11,500 rows, each with 178 sequential data points concatenated from the original dataset of 5 data folders, each including 100 files of EEG recordings of 23.6 seconds and containing 4097 data points. Each folder contained a single, original subset. Subset A contained EEG data gathering during epileptic seizure…. Subset B contained EEG data from brain tumor sites. Subset 3, from a healthy site where tumors had been located. Subsets 4 and 5 from non-seizure patients at rest with eyes open and closed, respectively.
Based on the described data, our team recognized that a Recurrent Neural Network (RNN) was needed to input the sequential data and return an output of whether the sequential data was a seizure or not. However, we realized that RNN models are known to get substantially large over time, reducing computation speeds. To help provide a solution to this issue, our group decided to implement a long-short-term memory (LSTM) model. After deciding our model’s architecture, we proceeded to train our model in two different DL frameworks inside Python, TensorFlow, and PyTorch. Through various rounds of retesting and redesigning, we were able to train and develop two accurate models in each of the models that not only performed well while learning the data while training, but also could accurately predict new data in the testing set (98 percent accuracy on the unseen data). These LSTM networks could classify normal EEG data when the brain waves are normal, and then immediately predict the seizure data based on if a dramatic spike occurred in the data.
After training our model, we had to implement our model in a real-life prototype scenario in which we utilized a Single Board Computer (SBC) in the Raspberry Pi 4 and a live capturing EEG headset in the Muse 2 Headband. The two hardware components would sync up through Bluetooth and the headband would return EEG data to the Raspberry Pi, which would process the data. Through the Muselsl API in Python, we were able to retrieve this EEG data in a format similar to the manner implemented during training. This new input data would be fed into our LSTM network (TensorFlow was chosen for the prototype due to its better performance than the PyTorch network), which would then output the result of the live captured EEG data in small intervals. This constant cycle would be able to accurately predict a seizure as soon as it occurs through batches of EEG data being fed into the LSTM network. Part of the reason why our research group chose the Muse Headband, in particular, was not only due to its compatibility with Python but also due to the fact that it was able to represent seizure data. Because none of our members had epilepsy, we had to find a reliable way of testing our model to make sure it worked on the new data. Through electrical disruptions in the wearable Muse Headband, we were able to simulate these seizures that worked with our network’s predictions. In our program, we implemented an alert system that would email the patient’s doctor as soon as a seizure was detected.

Individual wearing the Muse 2 Headband
Image Source: https://www.techguide.com.au/reviews/gadgets-reviews/muse-2-review-device-help-achieve-calm-meditation/
Sources Cited:
Wu, Q. & Fokoue, E. (2021). Epileptic seizure recognition data set: Data folder & Data set description. UCI Machine Learning Repository: Epileptic Seizure Recognition. Jan. 30. Center for Machine Learning and Intelligent Systems, University of California Irvine.
Nayak, C. S. (2020). EEG normal waveforms.” StatPearls [Internet]. U.S. National Library of Medicine, 31 Jul. 2020, www.ncbi.nlm.nih.gov/books/NBK539805/#.
Epilepsy. (2019). World Health Organization Fact Sheet. Jun. https://www.who.int/ news-room/fact-sheet s/detail/epilepsy
Other Related Articles published in this Open Access Online Scientific Journal include the following:
Developing Deep Learning Models (DL) for Classifying Emotions through Brainwaves
Reporter: Abhisar Anand, Research Assistant I
Machine Learning (ML) in cancer prognosis prediction helps the researcher to identify multiple known as well as candidate cancer diver genes
Curator and Reporter: Dr. Premalata Pati, Ph.D., Postdoc
Deep Learning-Assisted Diagnosis of Cerebral Aneurysms
Reporter: Dror Nir, PhD
Developing Machine Learning Models for Prediction of Onset of Type-2 Diabetes
Reporter: Amandeep Kaur, B.Sc., M.Sc.
Deep Learning extracts Histopathological Patterns and accurately discriminates 28 Cancer and 14 Normal Tissue Types: Pan-cancer Computational Histopathology Analysis
Reporter: Aviva Lev-Ari, PhD, RN
A new treatment for depression and epilepsy – Approval of external Trigeminal Nerve Stimulation (eTNS) in Europe
Reporter: Howard Donohue, PhD (EAW)
Mutations in a Sodium-gated Potassium Channel Subunit Gene related to a subset of severe Nocturnal Frontal Lobe Epilepsy
Reporter: Aviva Lev-Ari, PhD, RN
