Myc and Cancer Resistance
Curator: Larry H. Bernstein, MD, FCAP
Myc (c-Myc) is a regulator gene that codes for atranscription factor. The protein encoded by this gene is a multifunctional, nuclear phosphoprotein that plays a role in cell cycle progression, apoptosis and cellular transformation.[1]
Myc gene was first discovered in Burkitt lymphoma patients. In Burkitt lymphoma, cancer cells showchromosomal translocations, in which Chromosome 8 is frequently involved. Cloning the break-point of the fusion chromosomes revealed a gene that was similar to myelocytomatosis viral oncogene (v-Myc). Thus, the newfound cellular gene was named c-Myc.
http://www.ncbi.nlm.nih.gov/gene/17869
Protein increases signals that protect cancer cells
Researchers have identified a link between the expression of a cancer-related gene and cell-surface molecules that protect tumors from the immune system

{kind=link}
The Myc protein, depicted here, is mutated in more than half of all human cancers. Petarg/Shutterstock
A cancer-associated protein called Myc directly controls the expression of two molecules known to protect tumor cells from the host’s immune system, according to a study by researchers at the Stanford University School of Medicine.
The finding is the first to link two critical steps in the development of a successful tumor: uncontrolled cell growth — when mutated or misregulated, Myc causes an increase in the levels of proteins that promote cell division — and an ability to outwit the immune molecules meant to stop it.
The study was published online March 10 inScience. Dean Felsher, MD, PhD, a professor of oncology and of pathology, is the senior author. The lead author is postdoctoral scholar Stephanie Casey, PhD. The work was conducted in collaboration with researchers at the University of Wurzburg.
“Our findings describe an intimate, causal connection between how oncogenes like Myc cause cancer and how those cancer cells manage to evade the immune system,” Felsher said.
‘Don’t eat me’ and ‘don’t find me’
One of the molecules is the CD47 protein, which researchers in the Stanford laboratory of Irving Weissman, MD, have discovered serves as a “don’t eat me” signal to ward off cancer-gobbling immune cells called macrophages. Weissman is the Virginia and D.K. Ludwig Professor for Clinical Investigation in Cancer Research and the director of Stanford’s Institute for Stem Cell Biology and Regenerative Medicine.
Nearly all human cancers express high levels of CD47 on their surfaces, and an antibody targeting the CD47 protein is currently in phase-1 clinical trials for a variety of human cancers.
The other molecule is a “don’t find me” protein called PD-L1, known to suppress the immune system during cancer and autoimmune diseases but also in normal pregnancy. It’s often overexpressed on human tumor cells. An antibody that binds to PD-L1 has been approved by the U.S. Food and Drug Administration to treat bladder and non-small-cell lung cancer, but it has been shown to be effective in the treatment of many cancers.
Dean Felsher
Programmed death-ligand 1 (): an inhibitory immune pathway exploited by cancer
![Image of PD-L1 binding to B7.1 and PD-1, deactivating T cell]](https://i0.wp.com/www.researchcancerimmunotherapy.com/images/pathways/pd-l1-hero.jpg?resize=328%2C184)
http://www.researchcancerimmunotherapy.com/images/pathways/pd-l1-hero.jpg
{kind=link}
In cancer, Myc a usual suspect
Researchers in Felsher’s laboratory have been studying the Myc protein for more than a decade. It is encoded by a type of gene known as an oncogene. Oncogenes normally perform vital cellular functions, but when mutated or expressed incorrectly they become powerful cancer promoters. The Myc oncogene is mutated or misregulated in over half of all human cancers.
In particular, Felsher’s lab studies a phenomenon known as oncogene addiction, in which tumor cells are completely dependent on the expression of the oncogene. Blocking the expression of the Myc gene in these cases causes the complete regression of tumors in animals.
In 2010, Felsher and his colleagues showed that this regression could only occur in animals with an intact immune system, but it wasn’t clear why.
“Since then, I’ve had it in the back of my mind that there must be a relationship between Myc and the immune system,” said Felsher.
Turning off Myc expression
Casey and Felsher decided to see if there was a link between Myc expression and the levels of CD47 and PD-L1 proteins on the surface of cancer cells. To do so, they investigated what would happen if they actively turned off Myc expression in tumor cells from mice or humans. They found that a reduction in Myc caused a similar reduction in the levels of CD47 and PD-L1 proteins on the surface of mouse and human acute lymphoblastic leukemia cells, mouse and human liver cancer cells, human skin cancer cells, and human non-small-cell lung cancer cells. In contrast, levels of other immune regulatory molecules found on the surface of the cells were unaffected.
I’ve had it in the back of my mind that there must be a relationship between Myc and the immune system.
In publicly available gene expression data on tumor samples from hundreds of patients, they found that the levels of Myc expression correlated strongly with expression levels of CD47 and PD-L1 genes in liver, kidney and colorectal tumors.
The researchers then looked directly at the regulatory regions in the CD47 and PD-L1 genes. They found high levels of the Myc protein bound directly to the promoter regions of both CD47 and PD-L1 in mouse leukemia cells, as well as in a human bone cancer cell line. They were also able to verify that this binding increased the expression of the CD47 gene in a human blood cell line.
Possible treatment synergy
Finally, Casey and Felsher engineered mouse leukemia cells to constantly express CD47 or PD-L1 genes regardless of Myc expression status. These cells were better able than control cells to evade the detection of immune cells like macrophages and T cells, and, unlike in previous experiments from Felsher’s laboratory, tumors arising from these cells did not regress when Myc expression was deactivated.
“What we’re learning is that if CD47 and PD-L1 are present on the surfaces of cancer cells, even if you shut down a cancer gene, the animal doesn’t mount an adequate immune response, and the tumors don’t regress,” said Felsher.
The work suggests that a combination of therapies targeting the expression of both Myc and CD47 or PD-L1 could possibly have a synergistic effect by slowing or stopping tumor growth, and also waving a red flag at the immune system, Felsher said.
“There is a growing sense of tremendous excitement in the field of cancer immunotherapy,” said Felsher. “In many cases, it’s working. But it’s not been clear why some cancers are more sensitive than others. Our work highlights a direct link between oncogene expression and immune regulation that could be exploited to help patients.”
The research is an example of Stanford Medicine’s focus on precision health, the goal of which is to anticipate and prevent disease in the healthy and precisely diagnose and treat disease in the ill.
Other Stanford co-authors of the paper are oncology instructor Yulin Li, MD, PhD; postdoctoral scholars Ling Tong, PhD, Arvin Gouw, PhD, and Virginie Baylot, PhD; former research assistant Kelly Fitzgerald; and undergraduate student Rachel Do.
The research was supported by the National Institutes of Health (grants RO1CA089305, CA170378, CA184384, CA105102, P50 CA114747, U56CA112973, U01CA188383, 1F32CA177139 and 5T32AI07290).
The pathway downregulates cytotoxic activity to maintain immune homeostasis
Under normal conditions, the inhibitory ligands and play an important role in maintaining immune homeostasis.1 and bind to specific receptors on T cells. When bound to their receptors, cytotoxic T-cell activity is downregulated, thereby protecting normal cells from collateral damage.1,2
![Image showing PD-L1 binding to B7.1 and PD-1 to deactivate T cells during immune response]](https://i0.wp.com/www.researchcancerimmunotherapy.com/images/pathways/pd-l1-cta-2.jpg?resize=282%2C263)
PD-L1
Broadly expressed in multiple tissue types, including hematopoietic, endothelial, and epithelial cells1,4
B7.1
Receptor expressed on activated T cells and dendritic cells3
PD-1
Receptor expressed primarily on activated T cells3
CONVERSELY, BINDS PRIMARILY TO 3
![Image showing PD-L1 binding to B7.1 and PD-1 to deactivate T cells during immune response]](https://i0.wp.com/www.researchcancerimmunotherapy.com/images/pathways/pd-l1-cta-2.jpg?resize=290%2C271)
PD-L2
Restricted expression on immune cells and in some organs, such as the lung and colon1,4,5
PD-1
Receptor expressed primarily on activated T cells3
Many tumors can exploit the PD-L1 pathway to inhibit the antitumor response
In cancer, the and pathways can protect tumors from cytotoxic T cells, ultimately inhibiting the antitumor immune response in 2 ways.1-3
- Deactivating cytotoxic T cells in the tumor microenvironment
- Preventing priming and activation of new T cells in the lymph nodes and subsequent recruitment to the tumor
MAY INHIBIT CYTOTOXIC ACTIVITY IN THE TUMOR MICROENVIRONMENT
Upregulation of can inhibit the last stages of the cancer immunity cycle by deactivating cytotoxic T cells in the tumor microenvironment.1
Activated T cells in the tumor microenvironment release interferon gamma.2
As a result, tumor cells and tumor-infiltrating immune cells overexpress .2
binds to T-cell receptors B7.1 and PD-1, deactivating cytotoxic T cells. Once deactivated, T cells remain inhibited in the tumor microenvironment.1,2
MAY INHIBIT CANCER IMMUNITY CYCLE PROPAGATION IN THE LYMPH NODES
overexpression can also inhibit propagation of the cancer immunity cycle by preventing the priming and activation of T cells in the lymph nodes.1-3
expression is upregulated on dendritic cells within the tumor microenvironment.2,3
–expressing dendritic cells travel from the tumor site to the lymph node.4
binds to B7.1 and PD-1 receptors on cytotoxic T cells, leading to their deactivation.3
http://www.researchcancerimmunotherapy.com/pathways/pd-l1-immune-evasion
The cancer immunity cycle characterizes the complex interactions between the immune system and cancer
The cancer immunity cycle describes a process of how one’s own immune system can protect the body against cancer. When performing optimally, the cycle is self-sustaining. With subsequent revolutions of the cycle, the breadth and depth of the immune response can be increased.1
STEPS 1-3: INITIATING AND PROPAGATING ANTICANCER IMMUNITY1
- Oncogenesis leads to the expression of neoantigens that can be captured by dendritic cells
- Dendritic cells can present antigens to T cells, priming and activating cytotoxic T cells to attack the cancer cells
STEPS 4-5: ACCESSING THE TUMOR1
- Activated T cells travel to the tumor and infiltrate the tumor microenvironment
STEPS 6-7: CANCER-CELL RECOGNITION AND INITIATION OF CYTOTOXICITY1
- Activated T cells can recognize and kill target cancer cells
- Dying cancer cells release additional cancer antigens, propagating the cancer immunity cycle

http://www.researchcancerimmunotherapy.com/pathways/pd-l1
REFERENCES
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1-10. PMID: 23890059
- Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy—inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 2012;18:6580-6587. PMID: 23087408
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677-704. PMID: 18173375
- Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61-73. PMID: 23890064
Stephanie C. Casey1, Ling Tong1, Yulin Li1, Rachel Do1,…, Ines Guetegemann1, Martin Eilers2,3, Dean W. Felsher1,*
MYC Helps Cancer Hide
The transcriptional regulator dampens the immune system’s ability to elicit an antitumor response, a study shows.
| March 10, 2016 http://www.the-scientist.com/?articles.view/articleNo/45545/title/MYC-Helps-Cancer-Hide/

Model showing regulation of immunological checkpoints in MYC-driven tumors S.C. CASEY ET AL., SCIENCE
Myc, a transcriptional regulator that is overexpressed in several human cancers, appears to have a direct role in preventing immune cells from efficiently attacking tumor cells. The oncogene in part sustains tumor growth by increasing the levels of two immune checkpoint proteins, CD47 and programmed death-ligand 1 (PD-L1), which help thwart the host immune response, according to a study published today (March 10) in Science.
“It’s been shown that MYC is deeply involved in tailoring the external environment of proliferating tumor cells,” said Gerard Evan, a cancer researcher at the University of Cambridge, U.K., who was not involved in the study. “What is interesting here [is that MYC] tailors the ability of T cells to come in and survey the expanding tissue.”
“This study suggests . . . that MYC can drive expression of immune evasion molecules in cancer cells,” wrote Thomas Gajewski, a cancer researcher at the University of Chicago who was not involved in the study. “This is a novel result that could have translational implications if Myc-targeted drugs are found to be effective in the clinic.”
Combination strategies to enhance antitumor ADCC
The clinical efficacy of monoclonal antibodies as cancer therapeutics is largely dependent upon their ability to target the tumor and induce a functional antitumor immune response. This two-step process of ADCC utilizes the response of innate immune cells to provide antitumor cytotoxicity triggered by the interaction of the Fc portion of the antibody with the Fc receptor on the immune cell. Immunotherapeutics that target NK cells, γδ T cells, macrophages and dendritic cells can, by augmenting the function of the immune response, enhance the antitumor activity of the antibodies. Advantages of such combination strategies include: the application to multiple existing antibodies (even across multiple diseases), the feasibility (from a regulatory perspective) of combining with previously approved agents and the assurance (to physicians and trial participants) that one of the ingredients – the antitumor antibody – has proven efficacy on its own. Here we discuss current strategies, including biologic rationale and clinical results, which enhance ADCC in the following ways: strategies that increase total target–monoclonal antibody–effector binding, strategies that trigger effector cell ‘activating’ signals and strategies that block effector cell ‘inhibitory’ signals.
Monoclonal antibodies (mAbs) can target tumor antigens on the surface of cancer cells and have a favorable toxicity profile in comparison with cytotoxic chemotherapy. Expression of tumor antigens is dynamic and inducible through agents such as Toll-like receptor (TLR) agonists, immunomodulatory drugs (IMiDs) and hypomethylating agents [1]. Following binding of the mAb to the tumor antigen, the Fc portion of the mAb interacts with the Fc receptor (FcR) on the surface of effector cells (i.e., NK cells, γδ T cells and macrophages), leading to antitumor cytotoxicity and/or phagocytosis of the tumor cell. FcR interactions can be stimulatory or inhibitory to the killer cell, depending on which FcR is triggered and on which cell. Stimulatory effects are mediated through FcγRI on macrophages, dendritic cells (DCs) and neutrophils, and FcγRIIIa on NK cells, DCs and macrophages. In murine models, the cytotoxicity resulting from FcR activation on a NK cell, γδ T cell and macrophage is responsible for antitumor activity [2]. The role of DCs should be noted: although not considered to be primary ADCC effector cells, they can respond to mAb-bound tumor cells via their own FcR-mediated activation and probably play a significant role in activating effector cells. Preclinical models have shown that, although not the effector cell, DCs are critical to the efficacy of mAb-mediated tumor elimination [3]. Equally, mAb-activated ADCC effector cells can induce DC activation [4] and the importance of this crosstalk is an increasing focus of study [5].
The antitumor effects of mAbs are caused by multiple mechanisms of action, including cell signaling agonism/antagonism, complement activation and ligand sequestration, although ADCC probably plays a predominant role in the efficacy of some mAbs. In a clinical series, a correlation between the affinity of the receptor FcγRIIIa (determined by inherited FcR polymorphisms) and the clinical response to mAb therapy, supporting the significance of the innate immune response [6–10]. Several strategies could potentially improve the innate response following FcR activation by a mAb (Figure 1):
Quantitatively increasing the density of the bound target, mAb or the effector cells;
Stimulation of the effector cell by targeting the NK cell, γδ T cell and/or macrophage with small molecules, cytokines or agonistic antibodies;
Blocking an inhibitory interaction between the NK cell or macrophage and the tumor cell.

http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3386352/bin/nihms384451f1.jpg
{kind=link}
Enhancing ADCC
FcR: Fc receptor; HDACi: Histone deacetylase inhibitor; IMiD: Immunomodulator; KIR: Killer immunoglobulin-like receptor;
The ability of the combination approaches to enhance ADCC is largely determined by the capacity of the mAb to induce ADCC. Since the approval of the first mAb for the treatment of non-Hodgkin’s lymphoma, rituximab (RTX), in 1997, several mAbs have become standard of care for the treatment of both solid tumors and hematologic malignancies, including trastuzumab (TRAST), alemtuzumab, cetuximab, panitumumab and ofatumumab [11]. As noted above, clinical series among lymphoma patients treated with an anti-CD20 mAb (RTX) [6,7], HER2-expressing breast cancer receiving anti-HER2 mAb therapy (TRAST) [8] or colorectal cancer patients treated with an anti-EGFR mAb (cetuximab) [9,10] observed a correlation between clinical benefit and FcγRIIIa genotype, with patients who have higher-affinity polymorphisms demonstrating superior clinical outcomes. By contrast, the anti-EGFR mAb panitumumab does not induce ADCC, owing to a different Fc isotype that does not bind to the FcγRIIIa. Therefore, when considering enhancement of ADCC, such approaches are limited to combinations with mAbs that activate the FcR. Nonetheless, an advantage of this dual therapy strategy is that mAbs yet to be discovered against currently unknown tumor antigens may be combined with the therapeutics discussed herein.
Increasing target–mAb–effector binding
As the central element in the target–mAb–effector cell unit, the mAb seems to be a probable candidate for improvements, either in its antigen-binding or its Fc-binding domains. This approach has been heavily pursued with some degree of success [12–15]. Antibody engineering to improve interaction between the target or FcR requires that each new antibody be individually developed and tested as a new entity.
Increasing the antigen target
Tumor cells with a lower density of antigen targets are less responsive to mAbs than higher antigen-expressing diseases [16]. Therefore, it seems logical to try to increase the expression of the target on tumor cells. Antigen expression can be upregulated by cytokines [17], ionizing radiation [18], natural metabolites [19] and hypomethylating agents such as decitabine [20]. In addition, the family of TLR9 agonists known as CpG oligodeoxynucleotides (CpG ODN) can induce CD20 expression on malignant B cells [21–23]. Taken together with data showing the activating effect of CpG ODN on effector cells (discussed below), it seems reasonable that the combination of CpG ODN with mAb might have synergistic efficacy. Clinical series, however, have tested CpG ODN administered intravenously or subcutaneously and have observed little efficacy in Phase I and II studies [24–26] in low-grade lymphoma. One possible limitation of these studies has been their application to diseases (primarily follicular and mantle cell lymphoma) known to already have high expression of the relevant antigen (CD20). It is plausible that increasing antigen expression on low antigen-expressing diseases such as chronic lymphocytic leukemia could have a greater increase in relative efficacy. To this end, monotherapy studies have recently been undertaken [27,301] and should lead to combination trials.
……
Effector cells: γδ T cells
The role of NK cells and macrophages in mediating ADCC has been well established; however, only recently have γδ T cells been found to play a role as ADCC effectors. Typically, this population is considered as a minor subset (<5% of circulating T cells), although they may infiltrate tumors of epithelial origin preferentially and constitute a large portion of the tumor-infiltrating lymphocytes in cancers such as breast carcinoma. The combination of HLA-unrestricted cytotoxicity against multiple tumor cell lines of various histologies, secretion of cytolytic granules and proinflammatory cytokines such as TNF-α, IL-17 and IFN-γ make γδ T cells potentially potent antitumor effectors [32,33].
……
TLR agonists
In addition to its aforementioned induction of CD20, CpG ODN also indirectly augments innate immune function. TLRs are specialized to recognize pathogen-associated molecular patterns; they stimulate plasmacytoid DCs and B cells [53], and one of many plasmacytoid DC responses to stimulation by CpG ODNs is activation of local NK cells, thus improving spontaneous cytotoxicity and ADCC [54]. CpG ODN effects on NK cells appeared to be indirect and IFN-γ production by T cells (possibly in response to plasmacytoid DC activation) has been hypothesized as the intermediary of NK cell activation.
…..
Immunomodulatory drugs
IMiDs have shown clinical activity in multiple hematologic malignancies despite their primary mechanism of action being unclear. Among their biologic effects (particularly lenalidomide) there are demonstrable and pleiotropic effects on immune cells and signaling molecules. These include enhancement of in vitro NK cell- and monocyte-mediated ADCC on RTX-coated [68] as well as TRAST- and cetuximab-coated tumor cells [69]. In vivo studies in a human lymphoma severe combined immune deficiency mouse model demonstrated significant increases in NK cell recruitment to tumors mediated via microenvironment cytokine changes and augmented RTX-associated ADCC [70]. Studies suggest that IMiD activation of NK cells occurs indirectly; partly via IL-2 induction by T cells [71]. Clinically, a recent study noted significant increases in peripheral blood NK cells, NK cell cytotoxicity and serum IL-2, IL-15 and GM-CSF [72], the potential ADCC-promoting effects of which are discussed below.
…..
PD-1
PD-1 is a negative regulatory member of the CD28 superfamily expressed on the surface of activated T cells, B cells, NK cells and macrophages, similar to but more broadly regulatory than CTLA-4. Its two known ligands, PD-L1 and PD-L2, are both expressed on a variety of tumor cell lines. The PD-1–PD-L1 axis modulates the NK cell versus multiple myeloma effect, as seen by its blockade enhancing NK cell function against autologous primary myeloma cells, seemingly through effects on NK cell trafficking, immune complex formation with myeloma cells and cytotoxicity specifically toward PD-L1(+) tumor cells [179]. Two anti-PD-1 mAbs (BMS-936558 and CT-011) are currently in clinical trials, the latter in a combination study with RTX for patients with low-grade follicular lymphoma [314].
ConclusionThe recent approval of an anti-CTLA4 mAb has demonstrated that modulating the immune response can improve patient survival [180,181]. As the immune response is a major determinant of mAb efficacy, the opportunity now exists to combine mAb therapy with IMiDs to enhance their antitumor efficacy. Remarkable advances in the basic science of cellular immunology have increased our understanding of the effector mechanisms of mAb antitumor efficacy. Whereas the earliest iterations of such combinations, for example IL-2 and GM-CSF, may have augmented both effector and suppressive cells, newer approaches such as IL-15 and TLR agonists may more efficiently activate effector cells while minimizing the influence of suppressive cells. Despite these encouraging rationale and preliminary data, clinical evidence is still required to demonstrate whether combination therapies will increase the antitumor effects of mAb.
Still, this approach is unique in combining a tumor-targeting therapy, the mAb, with an immune-enhancing therapy. If successful, these therapies may be combined with multiple mAbs in routine practice, as well as novel mAbs yet to be developed. Various approaches including augmenting antigen expression, stimulating the innate response and blocking inhibitory signals are being explored to determine the optimal synergy with mAb therapies. Therapies targeting NK cells, γδ T cells, macrophages and DCs may ultimately be used in combination to further augment ADCC. Encouraging preclinical studies have led to a number of promising therapeutics, and the results of proof-of-concept clinical trials are eagerly awaited.
PD-L1, other targeted therapies await more standardized IHC
February 2016—Immunohistochemistry is heading down a path toward more standardization, and that’s essential as it plays an increasing role in rapidly expanding immunotherapy, says David L. Rimm, MD, PhD, professor of pathology and of medicine (oncology) and director of translational pathology at Yale University School of Medicine. As a co-presenter of a webinar produced by CAP TODAY in collaboration with Horizon Diagnostics, titled “Immunohistochemistry Through the Lens of Companion Diagnostics” (http://j.mp/ihclens_webinar), he analyzes the core challenges of IHC’s adaptation to the needs of precision medicine: binary versus continuous IHC, measuring as opposed to counting or viewing by the pathologist, automation, and assay performance versus protein measurement.
“Immunohistochemistry is 99 percent binary already,” Dr. Rimm points out. “There are only a few assays in our labs—ER, PR, HER2, Ki-67, and maybe a few more—where we really are looking at a continuous curve or a level of expression.”
Two criteria in the 2010 ASCO/CAP guidelines on ER and PR testing in breast cancer patients are key, he says: 1) the percentage of cells staining and 2) any immunoreactivity. “The first is hard to estimate, but the guidelines recommend the use of greater than or equal to one percent of cells that are immunoreactive. That means they could have a tiny bit of signal or they could have a huge amount of signal and they would be considered immunoreactive, which thereby makes this a binary test.”
Having the test be binary can be a problem for companion diagnostic purposes because any immunoreactivity is dependent on the laboratory threshold and counterstain. For example, if two of the same spots, serial sections on a tissue microarray, were shown side by side, one with and one without the hematoxylin counterstain, “you might see the counterstain make this positive test into a negative by eye, which is a potential problem with IHC when you have a binary stain.” (Fig. 1).

http://www.captodayonline.com/wordpress/wp-content/uploads/2016/02/Fig1.jpg
{kind=link}
Dr. Rimm describes a small study done with three different CLIA-certified labs, each using a different FDA-approved antibody and measuring about 500 breast cancer cases on a tissue microarray. The study showed there can be fairly significant discordance between labs—between 18 and 30 percent discordance—in terms of the cases that were positive. “In fact, if we look at outcome, 18 percent of the cases were called positive in Lab Two but were negative in Lab Three. Lab Three showed outcomes similar to the double positives whereas Lab Two had false-negatives.” This is an important problem that occurs when we try to binarize our immunohistochemistry, he says.
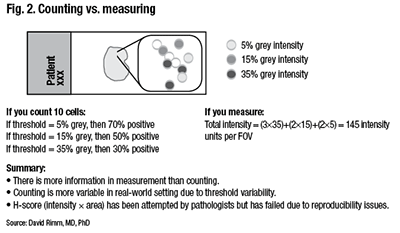
Counting is more variable in a real-world setting due to the variability of the threshold for considering a case positive. “You can easily calculate that if your threshold was five percent, then you’d have 70 percent positive cells. And you would easily call this positive. But if you added more hematoxylin because that’s how your pathologist liked it, then perhaps you’d only have 30 percent positive. So this is the risk of using thresholds.” (Fig. 2).

http://www.captodayonline.com/wordpress/wp-content/uploads/2016/02/Fig2.gif
{kind=link}
Although this is done in all of immunohistochemistry today, Dr. Rimm thinks it is an important consideration as IHC transitions to more standardized form. “An H score—intensity times area, which has been attempted many times, can’t be done by human beings. Pathologists try but have failed.”
“We can’t do those intensities by eye. We have to measure them with a machine. But we get a very different piece of information content when we measure intensity, as opposed to measuring the percentage of cells above a threshold. In sum, more information is present in a measurement than in counting.”
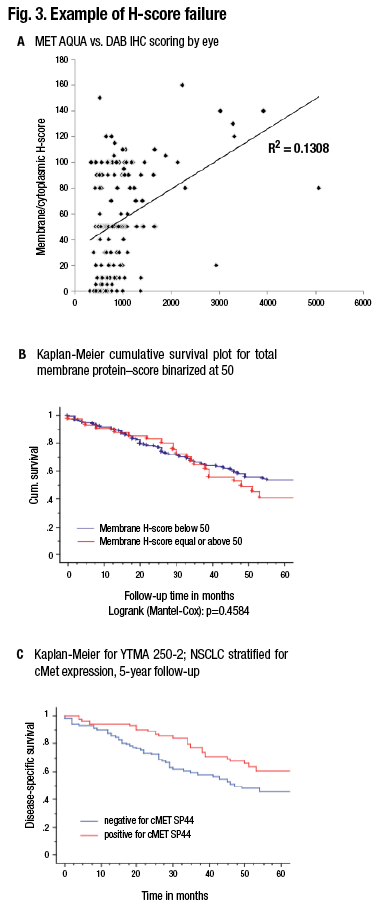
Pathologists read slides for a living, so it’s uncomfortable to think about giving that up in order to use a machine to measure the slides. “But I think if we want to serve our clients and our patients, we really owe them the accuracy of the 21st century as opposed to the methods of the 20th century.” (Fig. 3).

http://www.captodayonline.com/wordpress/wp-content/uploads/2016/02/Fig3.gif
{kind=link}
A shows comparison of a quantitative fluorescence score on the x axis versus an H-score on the y axis. Note the noncontinuous nature of human estimation of intensity times area (H-score). B) The survival curve in a population of lung cancer cases using the H-score. C) The survival curve in the same population using the quantitative score. (Source: David Rimm, MD, PhD)
Among the currently available quantitative measuring devices are the Visiopharm, VIAS (Ventana), Aperio (Leica), InForm (Perkin-Elmer), and Definiens platforms. “We use the platform invented in my lab, called Aqua [Automated Quantitative Analysis], but this is now owned by Genoptix/Novartis. Genoptix intends to provide commercial tests using Aqua internally,” Dr. Rimm says, “as well as enable platform and commercial testing through partnership with additional reference lab providers.
“There are many quantification platforms,” he adds, “and I believe that any of them, used properly, can be effective in measurement.”
(Of the 265 participants in the CAP PM2 Survey, 2015 B mailing, who reported using an imaging system for quantification, 4.6 percent use VIAS, 4.1 percent use ACIS, 0.8 use Applied Imaging, and 10 percent use “other” imaging systems. Of the 1,359 Survey participants who responded to the question about use of an imaging system to analyze hormone receptor slides, 1,094, or 80.5 percent, reported not using any imaging system for quantification.)
Says Dr. Rimm: “The first platform we used to try to quantitate some DAB stain slides was actually the Aperio Nuclear Image Analysis algorithm. But the problem with DAB is that you can’t see through it. And so inherently it’s physically flawed as a method for accurate measurement.” He compares DAB to looking at stacks of pennies from above, where their height and quantity can’t be surmised, as opposed to from the side, where their numbers can be accurately estimated. “This is why I don’t use, in general, DAB-type technologies or any chromogen.”
Fluorescence doesn’t have this problem, and that is the reason Dr. Rimm began using fluorescence as a quantitative method. “We try to be entirely quantitative without any feature extraction. So we define epithelial tumors using a mask of cytokeratin. We define a mask by bleeding and dilating, filling some holes, and then ultimately measure the intensity of each cell, or of each target we’re looking for. In this case, in a molecularly defined compartment.”
Compartments can be defined by any type of molecular interactions. “We defined DAPI-positive pixels as nuclei, and we measure the intensity of the estrogen receptor within the compartment. And that gives us an intensity over an area or the equivalent of a concentration.” Many other fluorescent tools can be used in this same manner, but he cautions against use of fluorescent tools that group and count. “That’s a second approach that can be used, but the result gives you a count instead of a measurement.”
When comparing a pathologist’s reading versus a quantitative immunofluorescence score, he notes, pathologists actually don’t generate a continuous score. Instead, pathologists tend to use groups. “We tend to use a 100 or a 200 or an even number. We never say, ‘Well, it’s 37 percent positive.’ We say, ‘It’s 40 percent positive,’ because we know we can’t reproducibly tell 37 from 38 from 40 percent positive.”
The result of that is a noncontinuous scoring result, which doesn’t give the information content of quantitative measurement. A comparison between the two methods shows that at times, where quantitative measurement shows a significant difference in outcome, nonquantitative measure or an H-score difference may not show a difference in outcome. (Fig. 3 illustrates this concept.)
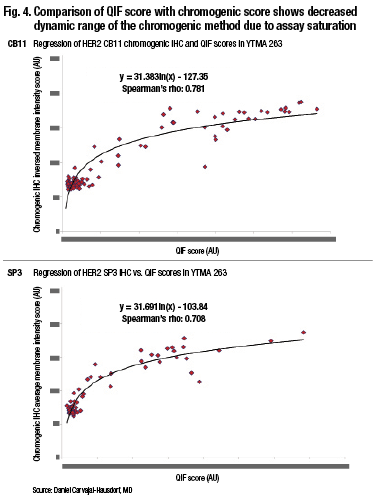
“Pathologists tend to group things, and we also tend to overestimate. It’s not that pathologists are bad readers. It’s just the tendency of the human eye because of our ability to distinguish different intensities and the subtle difference between intensities. But even if you compare two quantitative methods, you can see that the method where light absorbance occurs—that is the percent positive nuclei by Aperio, which is a chromogen-based method—tends to saturate. This is, in fact, amplified dramatically when you look at something with a wide dynamic range like HER2.” (Fig. 4).

http://www.captodayonline.com/wordpress/wp-content/uploads/2016/02/Fig4.gif
{kind=link}
In one study, researchers found less than one percent discordance—essentially no discordance—between two antibodies (Dekker TJ, et al. Breast Cancer Res. 2012;14[3]:R93). But looking at these results graphed quantitatively, you would see a very different result, Dr. Rimm says. “You can see a whole group of cases down below where there’s very low extracellular domain and very high cytoplasmic domain. In fact, some of these cases have essentially no extracellular domain, but high levels of cytoplasmic domain, and other cases have roughly equal levels of each” (Carvajal-Hausdorf DE, et al. J Natl Cancer Inst.2015;107[8]:pii:djv136).
Recent studies by Dr. Rimm’s group have shown this to have clinical implications. He looked at patients treated with trastuzumab in the absence of chemotherapy, in an unusual study called the HeCOG (Hellenic Cooperative Oncology Group) trial.
“We found that patients who had high levels of both extracellular and intracellular domain have much more benefit than patients who are missing the extracellular domain and thereby missing the trastuzumab binding site.” Follow-up studies are being done to validate this finding in larger cohorts.
Preanalytical variables, Dr. Rimm emphasizes, can have significant effects on IHC results, and more than 175 of them have been identified. “These are basically all the things we can’t control, which is the ultimate argument for standardization.”
In a surprising study by Flory Nkoy, et al., he says, it was shown that breast cancer specimens were more likely to be ER negative if the patient’s surgery was on a Friday because there was a higher ER-negative rate on Friday than on Monday. “So how could that be? Well, it was clearly the fact that the tissue was sitting over the weekend. And when it sat over the weekend, the ER positivity rate was going down” (Arch Pathol Lab Med. 2010;134:606–612).
Another study showed that after one hour, four hours, and eight hours of storage at room temperature, you lose significant amounts of staining, Dr. Rimm says. “And perhaps the best nonquantitative study or H-score-based study of this phenomenon was done by Isil Yildiz-Aktas, et al., where a significant decrease in the estrogen receptor score was found after only three hours in delay to fixation” (Mod Pathol. 2012;25:1098–1105).
How long the slide is left to sit after it is cut is another preanalytical variable to be concerned with. “In the clinical lab, that’s not often a problem since we cut them, then stain them right away. But in a research setting, a fresh-cut slide can look very different from a slide that’s two days old, six days old, or 30 days old, where a 2+ spot on a breast cancer patient becomes negative after 30 days sitting on a lab bench. So those are both key variables to be mindful of.”
One solution for those preanalytic variables is trying to prevent delayed time to fixation. “And probably time to fixation is one of the main preanalytic variables, although it’s only one of the many hundreds of variables. The method we use to try to get around this problem is to use core biopsies or allow rapid and complete fixation, and then other things can be done.”
Finally, he warns, don’t cut your tissue until right before you stain it. “If you’re asked to send a tissue out to a collaborator or someone who is going to use it for research purposes later, we recommend coring and re-embedding the core, or sending the whole block. Unstained sections, when not properly stored in a vacuum, will ultimately be damaged by hydration or oxidation, both of which lead to loss of antigenicity.”
The crux of the matter is assay performance versus protein measurement, Dr. Rimm says. “In the last six to nine months, we really are faced with this problem in spades, as PD-L1 has become a very important companion diagnostic.”
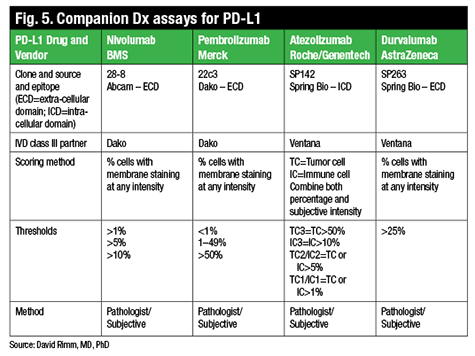
There are now four PD-L1 drugs with complementary or companion diagnostic tests (Fig. 5). One of the FDA-approved drugs, nivolumab (Opdivo, Bristol-Myers Squibb), for example, uses a clone called 28-8, which is provided by Dako in an assay, a complementary diagnostic assay, and with the following suggested scoring system: one percent, five percent, or 10 percent. In contrast, pembrolizumab (Keytruda, Merck) is also now FDA-approved but requires a companion diagnostic test that uses a different antibody, although the same Dako Link 48 platform. This diagnostic has a different scoring system of less than one percent, one to 49 percent, and 50 percent and over.
http://www.captodayonline.com/wordpress/wp-content/uploads/2016/02/Fig5.gif
{kind=link}
Two other companies, Roche/Genentech and AstraZeneca, also have drugs in trials that may or may not have companion diagnostic testing, though both have already identified a partner and a unique antibody (neither of those listed above) and companion diagnostic testing scores used in their clinical trials.
“So what’s a pathologist to do?” Dr. Rimm says. “Well, there are a few problems with this. First of all, what we really should be doing is measuring PD-L1. That’s the target and that’s what should ultimately predict response. But instead what we’re stuck with, through the intricacies of the way our field has grown and our legacy, is closed-system assays. While these probably do measure PD-L1, we do not know how these compare to each other.” Two parallel large multi-institutional studies are addressing this issue now, he says.
There are solutions for managing these closed-system assays to be sure the assay is working in your lab and that you can get the right answer, Dr. Rimm says. His laboratory uses a closed-system assay for PD-L1, relying not on the defined system but rather on a test system it has developed in doing a study with different investigators.
Sample runs by these different investigators show the potentially high variability, he says. “In a scan of results, no one would deny which spots are the positive spots and which are the negative.” But the difference in staining prevents accurate measurement of these things and shows the variability inherent even in a closed-box system.
A comparison of two closed-box systems, the SP1 run on the Discovery Ultra on Ventana, and the SP1, same antibody, run on the Dako closed-box system, also shows that, in fact, there’s not 100 percent agreement using same-day, same-FDA-cleared antibody staining and different autostainers. So automation may not solve the problem, Dr. Rimm notes (Fig. 6).
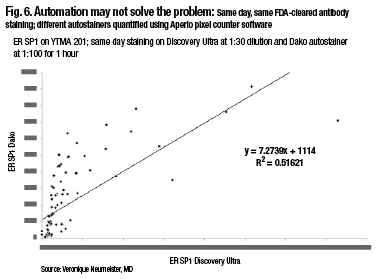
“When running these in a quantitative fashion and measuring them quantitatively, there are actually differences in the way these closed-box systems run. And so you, as the pathologist, have to be the one who makes sure your assays are correct, your thresholds are correct, and your measurements are accurate.”
The way to do that, he believes, is to use standardization or index arrays. An index array of HER2 that his laboratory developed has 3+ amplified, 2+ amplified, not amplified, and so on from 80 cases in the lab’s archive, shown stained with immunofluorescence and quantitative and DAB stain. “It was only with this standardization array, run every time we ran our stainer, that we were able to draw the conclusions in the previous study about extracellular versus cytoplasmic domain.”
Companies have realized the importance of this, and specifically companies like NantOmics (formerly OncoPlexDx) have realized they can exactly quantitate the amount of tissue on a slide using a specialized mass spectrometry method, he says. “They can actually give you amol/µg of total protein.”
He and colleagues are working with NantOmics now to try to convert from amols to protein to average quantitative fluorescent scores to help build these standards and make standard arrays more accurate. “This is still a work in progress, but I believe this is ultimately the kind of accuracy that can standardize all of our labs. We have shown that the quantitative fluorescence system is truly linear and quantitative for EGFR measurements when using mass spectrometry as a gold standard.” They are preparing to submit a manuscript with this data.
In the interim, Dr. Rimm’s laboratory has begun working also with Horizon Diagnostics, employing Horizon’s experimental 15-spot positive-control array. “When you use this array and quantitate it with quantitative fluorescence, you get a very interesting profile. If a cut point is set at one point, you would see three clearly positive cells or spots and 12 clearly negative spots with two different antibodies. But is that the threshold?”
“In fact, using a little higher score and a very quantitative test, you might find that the threshold may, in fact, be a little bit lower than that.” It turns out that only three of these 12 spots are true negatives. The others at least have some level of RNA, and some have a lot. “So how do we handle these? And are these behaving the same way with multiple antibodies?” Parallel results, finding nearly the same threshold case, have been found using SP142 from Ventana, E1L3N from Cell Signaling, and SP263 from Ventana.
Studies to address those issues are still in the early stage, he says. He cautions that there is variance in these assays, and more work is being done to reproduce the data. “But I think the important point is that, using these kinds of arrays, you can definitively determine whether your lab has the same cut point as every other lab. And were we to quantitate this with mass spectrometry, we would know exactly the break point for use in the future.”
Dr. Rimm’s laboratory has also built its own PD-L1 index tissue microarray with a number of its own tumor slides ranging from very low to very high expressors, a series of cell lines, and including some placenta-positive controls on normal tumor. He has found that generating an index array has advantages, and he encourages other laboratories to prepare their own index arrays to increase the accuracy and reproducibility of their laboratory-developed tests. “You can produce these in your own lab so that you can be sure you can standardize your tests run in your clinical lab from day to day and week to week as part of an LDT.”
“If we think about it, there really are no clinical antibodies today that are truly quantitative,” Dr. Rimm says. “And when there are, new protocols will be required, but I believe those protocols are now in existence. We just await the clinical trials that require truly quantitative protein measurement or in situ proteomics.”
In that process of moving toward in situ proteomics, suggests web-inar co-presenter Clive Taylor, MD, DPhil, professor of pathology in the Keck School of Medicine at the University of Southern California, FDA approval, per se, will not solve any of the problems discussed in the webinar. (See the January 2016 issue for the full report of Dr. Taylor’s presentation.) “I think what the FDA approval will do is demand that we find solutions to these problems ourselves. The FDA’s attitude is, to a large degree, dependent on the claim. So if we just use immunohistochemistry as a simple stain, then the FDA classes that as sort of class I, level 1. And we can do that [IHC stain] without having to get preapproval by the FDA.
“On the other hand, if we take something like the well-established HercepTest, where based on the result of that test alone, it’s decided whether or not the patient gets treatment, treatment that’s very expensive and treatment that has benefits and…side effects. That claim is, in fact, a very high-level claim. And for that, the FDA is demanding high-level data, which I think is entirely appropriate,” Dr. Taylor says.
Most of these upcoming companion diagnostics, if not all, he says, will be regarded by the FDA as class III, high level or high complexity. They will require a premarket approval study in conjunction with a clinical trial. And the FDA will demand high standards of control and performance, eventually. “There are not many labs that can produce those high standards as in-house or lab-developed tests today. And even the companies currently in trials are not producing the improved performance level for these tests that we are talking about today, as being required for high-quality quantitative and reproducible companion diagnostics. Eventually, I am convinced we will have to do that. It’s just that it will take time to get there.”
The FDA can only approve what is brought to it, Dr. Rimm points out. And so a true, fully quantitative IHC-based assay has presumably never been submitted, or at least never been approved by the FDA. “What we’re seeing instead are the assays that the FDA has approved, which are well defined and rigorously submitted. However, the result is a closed system that we use, which may or may not accurately measure PD-L1 on the slide, depending upon preanalytic variables and individual laboratories’ methods.”
“So questions keep popping up. And I can only say that we, as pathologists, have the final responsibility to our patients. And while it may not be recommended and it may change in the future, right now lab-derived tests or LDTs may be more accurate than FDA-approved platforms.”
“If you think about it, in molecular diagnostics where I’m familiar with EFGR and BRAF and KRAS tests, in that testing setting, less than 25 percent of the labs that do that test actually use the FDA-approved test,” Dr. Rimm says. “The remainder of the labs do their own LDTs, including our labs here at Yale.”
It wouldn’t surprise him if the same thing happens for PD-L1. “I’m aware of at least two labs—and we probably will be the third—that devise our own LDT for PD-L1 testing using the standards I’ve discussed, using array-type controls to be sure that our levels are correct, and then using a scoring system that we derived.”
“We aren’t really in a position to know at the time that we receive a piece of lung cancer tissue whether the oncologist is going to use pembrolizumab, which requires a companion diagnostic, or nivolumab, or the other drugs, which may or may not require a companion diagnostic. So in that sense, we’re almost bound to use an LDT,” Dr. Rimm says, since his lab can’t actually run four different potentially incongruent, though FDA-approved, tests for PD-L1.
Until a truly quantitative approach is developed and submitted to the FDA and approved, Dr. Taylor believes we won’t see things changing. “The algorithms that currently are approved have been approved on the basis that they can produce a similar result to a consensus group of pathologists. So they’re only as good as the pathologists.”
“As Dr. Rimm has discussed, I actually believe we can get a much better result than the pathologists can get with their naked eye. We have to get away from comparing it to what we currently can do and start to try to construct a proper test, just like we did in the clinical lab 30 years ago when we automated the clinical lab,” Dr. Taylor says. “We need to automate anatomic pathology, including the sample preparation, the assay process, and the reading, all three together in a closed system. And we’re nibbling away at the edges of it. We’ll get there, but it’ll take some time.”
Dr. Rimm is skeptical that the diagnostics field has learned any lessons from HercepTest and the companion diagnostics world of almost 20 years ago. “The submissions to the FDA for PD-L1 look very similar to what was submitted in 1998 for the HercepTest, the companion diagnostic test for trastuzumab [Herceptin]. And that’s disappointing. I think that is 20-year-old technology and we can do better. But even if we want to use the 20- or 40-year-old DAB-based technology, we should still be standardizing it and having a mechanism for standardization and having defined thresholds.”
As future FDA submissions come in, Dr. Rimm hopes that “even if they’re not quantitated, they can be standardized as to where the thresholds occur, so that we can be sure we deliver the best possible care to patients. And in the interim, I think we, as pathologists, will have to do that standardization with an LDT to be sure we’re giving our best results.”
Dr. Taylor warns that there is only a limited number of labs in the country and in the world that will be able to produce these LDTs, because of the complexity. “The FDA has already said in a position paper that it believes it may have to regulate LDTs to some extent. And what that will mean is that in the validation process, your own LDT will start to approach what is required for an FDA-approved test. And most labs are in no position to be able to do that.”
“So I think we’re going to come to a blending here, all forced by companion diagnostics. This is in situ proteomics,” Dr. Taylor says. “It’s a new test, essentially. It’s not straightforward immunohistochemistry, but a new test. And I think the fluorescence approach that Dr. Rimm has used has a lot of advantages in relating signal to target in terms of figure out what the best test is and stop comparing it to the pathologists. We should compare it to the best assay we can produce.”
With respect to the PD-L1 problem, Dr. Rimm notes, “I would point out that there is a so-called ‘Blueprint’ for comparison of the different antibodies and the different FDA assays, or potentially FDA-submitted tests anyway, to see how equivalent they are.” Similarly, he adds, the National Comprehensive Cancer Network recently issued a press release describing a multi-institutional study to assess the FDA-approved assay but also including an LDT (the Cell Signaling antibody E1L3N using the Leica Bond staining platform).
He points to a newly published study by his group (McLaughlin J, et al. JAMA Oncol. 2016;2[1]:46–54), finding that objective determination of PD-L1 protein levels in non-small cell lung cancer reveals heterogeneity within tumors and prominent interassay variability or discordance. The authors concluded that future studies measuring PD-L1 quantitatively in patients treated with anti-PD-1 and anti PD-L1 therapies may better address the prognostic or predictive value of these biomarkers. With future rigorous studies, including tissues with known responses to anti-PD-1 and anti-PD-L1 therapies, researchers could determine the optimal assay, PD-L1 antibody, and the best cut point for PD-L1 positivity.
Other work that will probably come out in mid-2016 from Dr. Rimm’s group has shown that expression of PD-L1 is largely bimodal, he says. “That is, there’s a group of patients that express a lot, and then there’s another group of patients that expresses a little or none.”
So time will tell how PD-L1 will be scored. “But if you look at the data from the Merck study and their cut point of greater than 50 percent, or even the cut point from the AstraZeneca studies of greater than 25 percent, you’re really dichotomizing the population into patients who are truly PD-LI positive from patients who are negative or almost negative.”
“Of course, we don’t want to miss patients in that negative to almost-negative group who will respond,” Dr. Rimm says. “On the other hand, we probably will have fairly good specificity and sensitivity with the assay defined by Merck and Dako with 22C3 as was recently published” (Robert C, et al. N Engl J Med. 2015;372[26]:2521–2532).
Many difficulties lie ahead, as researchers try to weigh the merits of different drugs with different approved tests on different platforms, involving different antibodies, Dr. Taylor says. “Does the lab try to set up four different PD-L1s, and if we only have one platform and not another, what do we do about that?” He thinks the tests may often be sent out to larger reference labs or academic centers as a result.
Dr. Rimm confirms that his own lab’s LDT—although literally thousands of PD-L1 tests have been conducted using it—is not yet up and running in the Yale CLIA laboratory, and in the meantime the IHC slides are being sent out to a commercial vendor.
Eventually, Dr. Taylor believes, the pressure of these dilemmas will lead the diagnostics field to develop an immunoassay on tissue sections. “We’ve never been forced to do that before, but once we are, that will produce a huge change in diagnostic capability and research capability.”
Anti–PD-1/PD-L1 therapy of human cancer: past, present, and future
Lieping Chen and http://www.jci.org/articles/view/80011
The cDNA of programmed cell death 1 (PD-1) was isolated in 1992 from a murine T cell hybridoma and a hematopoietic progenitor cell line undergoing apoptosis (1). Genetic ablation studies showed that deficiencies in PD-1 resulted in different autoimmune phenotypes in various mouse strains (2, 3). PD-1–deficient allogeneic T cells with transgenic T cell receptors exhibited augmented responses to alloantigens, indicating that the PD-1 on T cells plays a negative regulatory role in response to antigen (2).
Several studies contributed to the discovery of the molecules that interact with PD-1. In 1999, the B7 homolog 1 (B7-H1, also called programmed death ligand-1 [PD-L1]) was identified independently from PD-1 using molecular cloning and human expressed-sequence tag database searches based on its homology with B7 family molecules, and it was shown that PD-L1 acts as an inhibitor of human T cell responses in vitro (4). These two independent lines of study merged one year later when Freeman, Wood, and Honjo’s laboratories showed that PD-L1 is a binding and functional partner of PD-1 (5). Next, it was determined that PD-L1–deficient mice (Pdl1 KO mice) were prone to autoimmune diseases, although this strain of mice did not spontaneously develop such diseases (6). It became clear later that the PD-L1/PD-1 interaction plays a dominant role in the suppression of T cell responses in vivo, especially in the tumor microenvironment (7, 8).
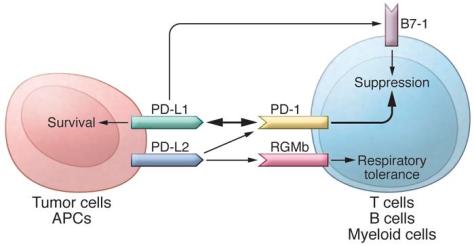
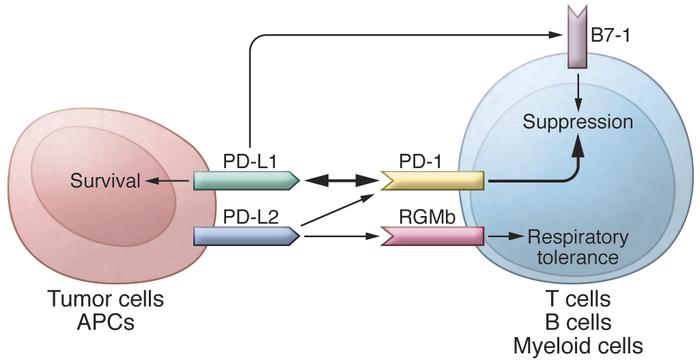
In addition to PD-L1, another PD-1 ligand called B7-DC (also known as PD-L2) was also identified by the laboratories of Pardoll (9) and Freeman (10). This PD-1 ligand was found to be selectively expressed on DCs and delivered its suppressive signal by binding PD-1. Mutagenesis studies of PD-L1 and PD-L2 molecules guided by molecular modeling revealed that both PD-L1 and PD-L2 could interact with other molecules in addition to PD-1 and suggested that these interactions had distinct functions (11). The functional predictions from these mutagenesis studies were later confirmed when PD-L1 was found to interact with CD80 on activated T cells to mediate an inhibitory signal (12, 13). This finding came as a surprise because CD80 had been previously identified as a functional ligand for CD28 and cytotoxic T lymphocyte antigen-4 (CTLA-4) (14, 15). PD-L2 was also found to interact with repulsive guidance molecule family member b (RGMb), a molecule that is highly enriched in lung macrophages and may be required for induction of respiratory tolerance (16). With at least five interacting molecules in the PD-1/PD-L1 pathway (referred to as the PD pathway) (Figure 1), further studies will be required to understand the relative contributions of these molecules during activation or suppression of T cells.
http://dm5migu4zj3pb.cloudfront.net/manuscripts/80000/80011/medium/JCI80011.f1.jpg
{kind=link}
The PD pathway.
The PD pathway has at least 5 interacting molecules. PD-L1 and PD-L2, with different expression patterns, were identified as ligands of PD-1, and the interaction of PD-L1 or PD-L2 with PD-1 may induce T cell suppression. PD-L1 was found to interact with B7-1 (CD80) on activated T cells and inhibit T cell activity. PD-L2 has a second receptor, RGMb; initially, this interaction activates T cells, but it subsequently induces respiratory tolerance. PD-L1 on tumor cells can also act as a receptor, and the signal delivered from PD-1 on T cells can protect tumor cells from cytotoxic lysis.
The discovery of the PD pathway did not automatically justify its application to cancer therapy, especially after the initial PD-1–deficient mouse studies, which suggested that PD-1 deficiency increases the incidence of autoimmune diseases (2, 3). In our initial work to characterize PD-L1 and its function, PDL1 mRNA was found to be broadly expressed in various tissues (17). However, normal human tissues seldom express PD-L1 protein on their cell surface, with the exception of tonsil (17), placenta (18), and a small fraction of macrophage-like cells in lung and liver (17), suggesting that, under normal physiological conditions, PDL1 mRNA is under tight posttranscriptional regulation. In sharp contrast, PD-L1 protein is abundantly expressed on the cell surface in various human cancers, as indicated by immunohistochemistry in frozen human tumor sections. Additionally, the pattern of PD-L1 expression was found to be focal rather than diffuse in most human cancers (17). In fact, the majority of in vitro–cultured tumor lines of both human and mouse origin are PD-L1–negative on the cell surface, despite overwhelming PD-L1 signal in specimens that are freshly isolated from patients with cancer (17, 19). This discrepancy was explained by the finding that IFN-γ upregulates PD-L1 on the cell surface of normal tissues and in various tumor lines (7, 17, 19). It was widely thought that IFN-γ typically promotes, rather than suppresses, T cell responses by stimulating antigen processing and presentation machinery (20, 21); therefore, the role of IFN-γ in downregulating immune responses in the tumor microenvironment via induction of PD-L1 was not well accepted until more recently. This finding is vital to our current understanding of the unique immunology that takes place in the tumor microenvironment and provided an important clue that led to the “adaptive resistance” hypothesis (see below) that explains this pathway’s mechanism of action to evade tumor immunity.
Due to the lack of cell surface expression of PD-L1 on most cultured tumor lines, it is necessary to reexpress PD-L1 on the surface using transfection to recapitulate the effects of cell surface PD-L1 in human cancers and to create models to study how tumor-associated PD-L1 interacts with immune cells. We now know that cancer cells and other cells in the tumor microenvironment can upregulate the expression of PD-L1 after encountering T cells, mostly via IFN-γ, which may make the transfection-mediated expression of PD-L1 unnecessary in some tumor models. Nevertheless, our results demonstrated that PD-L1+ human tumor cells could eliminate activated effector T cells (Teffs) via apoptosis in coculture systems, and this effect could be blocked by inclusion of an anti-human PD-L1 mAb (clone 2H1). Next, we generated a hamster mAb (clone 10B5) against mouse PD-L1 to block its interaction with T cells and test its role in tumor immunity in vitro and in vivo. We demonstrated that progressive growth of PD-L1+ murine P815 tumors in syngeneic mice could be suppressed using anti–PD-L1 mAb (17). Altogether, these studies represented the initial attempt at using mAb to block the PD pathway as an approach for cancer therapy. These proof-of-concept studies (17) were confirmed by several subsequent studies. A study from Nagahiro Minato’s laboratory showed that the J558L mouse myeloma line constitutively expressed high levels of cell surface PD-L1 and the growth of these cells in syngeneic BALB/c mice could be partially suppressed by administering anti–PD-L1 mAb (22). Our laboratory showed that regression of progressively growing squamous cell carcinomas in syngeneic mice could also be suppressed using a combination of adoptively transferred tumor-draining lymphocytes and anti–PD-L1 mAb (23). Furthermore, the Zou laboratory demonstrated that ovarian cancer–infiltrating human T cells could be activated in vitro using DCs, which showed enhanced activity in the presence of anti–PD-L1 mAb; upon transfer, these cells could eliminate established human ovarian cancers in immune-deficient mice (24). These early studies established the concept that the PD pathway could be used by tumors to escape immune attack in the tumor microenvironment. More importantly, these studies built a solid foundation for the development of anti-PD therapy for the treatment of human cancers. …..
Anti-PD therapy has taken center stage in immunotherapies for human cancer, especially for solid tumors. This therapy is distinct from the prior immune therapeutic agents, which primarily boost systemic immune responses or generate de novo immunity against cancer; instead, anti-PD therapy modulates immune responses at the tumor site, targets tumor-induced immune defects, and repairs ongoing immune responses. While the clinical success of anti-PD therapy for the treatment of a variety of human cancers has validated this approach, we are still learning from this pathway and the associated immune responses, which will aid in the discovery and design of new clinically applicable approaches in cancer immunotherapy.
PD-1 Pathway Inhibitors: Changing the Landscape of Cancer Immunotherapy
Dawn E. Dolan, PharmD, and Shilpa Gupta, MD
Background: Immunotherapeutic approaches to treating cancer have been evaluated during the last few decades with limited success. An understanding of the checkpoint signaling pathway involving the programmed death 1 (PD-1) receptor and its ligands (PD-L1/2) has clarified the role of these approaches in tumor-induced immune suppression and has been a critical advancement in immunotherapeutic drug development. Methods: A comprehensive literature review was performed to identify the available data on checkpoint inhibitors, with a focus on anti–PD-1 and anti–PD-L1 agents being tested in oncology. The search included Medline, PubMed, the ClinicalTrials.gov registry, and abstracts from the American Society of Clinical Oncology meetings through April 2014. The effectiveness and safety of the available anti–PD-1 and anti–PD-L1 drugs are reviewed. Results: Tumors that express PD-L1 can often be aggressive and carry a poor prognosis. The anti–PD-1 and anti–PD-L1 agents have a good safety profile and have resulted in durable responses in a variety of cancers, including melanoma, kidney cancer, and lung cancer, even after stopping treatment. The scope of these agents is being evaluated in various other solid tumors and hematological malignancies, alone or in combination with other therapies, including other checkpoint inhibitors and targeted therapies, as well as cytotoxic chemotherapy. Conclusions: The PD-1/PD-L1 pathway in cancer is implicated in tumors escaping immune destruction and is a promising therapeutic target. The development of anti–PD-1 and anti–PD-L1 agents marks a new era in the treatment of cancer with immunotherapies. Early clinical experience has shown encouraging activity of these agents in a variety of tumors, and further results are eagerly awaited from completed and ongoing studies.
……
Role of PD-1/PD-L1 Pathway PD-1 is an immunoinhibitory receptor that belongs to the CD28 family and is expressed on T cells, B cells, monocytes, natural killer cells, and many tumor-infiltrating lymphocytes (TILs)10; it has 2 ligands that have been described (PD-L1 [B7H1] and PD-L2 [B7-DC]).11 Although PD-L1 is expressed on resting T cells, B cells, dendritic cells, macrophages, vascular endothelial cells, and pancreatic islet cells, PD-L2 expression is seen on macrophages and dendritic cells alone.10 Certain tumors have a higher expression of PD-L1.12 PD-L1 and L2 inhibit T-cell proliferation, cytokine production, and cell adhesion.13 PD-L2 controls immune T-cell activation in lymphoid organs, whereas PD-L1 appears to dampen T-cell function in peripheral tissues.14 PD-1 induction on activated T cells occurs in response to PD-L1 or L2 engagement and limits effector T-cell activity in peripheral organs and tissues during inflammation, thus preventing autoimmunity. This is a crucial step to protect against tissue damage when the immune system is activated in response to infection.15-17 Blocking this pathway in cancer can augment the antitumor immune response.18 Like the CTLA-4, the PD-1 pathway down-modulates Tcell responses by regulating overlapping signaling proteins that are part of the immune checkpoint pathway; however, they function slightly differently.14,16 Although the CTLA-4 focuses on regulating the activation of T cells, PD-1 regulates effector T-cell activity in peripheral tissues in response to infection or tumor progression.16 High levels of CTLA-4 and PD-1 are expressed on regulatory T cells and these regulatory T cells and have been shown to have immune inhibitory activity; thus, they are important for maintaining self-tolerance.16 The role of the PD-1 pathway in the interaction of tumor cells with the host immune response and the PD-L1 tumor cell expression may provide the basis for enhancing immune response through a blockade of this pathway.16 Drugs targeting the PD-1 pathway may provide antitumor immunity, especially in PD-L1 positive tumors. Various cancers, such as melanoma, hepatocellular carcinoma, glioblastoma, lung, kidney, breast, ovarian, pancreatic, and esophageal cancers, as well as hematological malignancies, have positive PD-L1 expression, and this expression has been correlated with poor prognosis.8,19 Melanoma and kidney cancer are prototypes of immunogenic tumors that have historically been known to respond to immunotherapeutic approaches with interferon alfa and interleukin 2. The CTLA-4 antibody ipilimumab is approved by the US Food and Drug Administration for use in melanoma. Clinical activity of drugs blocking the PD-1/PD-L1 pathway has been demonstrated in melanoma and kidney cancer.20-24 In patients with kidney cancer, tumor, TIL-associated PD-L1 expression, or both were associated with a 4.5-fold increased risk of mortality and lower cancer-specific survival rate, even after adjusting for stage, grade, and performance status.18,19,25,26 A correlation between PD-L1 expression and tumor growth has been described in patients with melanoma, providing the rationale for using drugs that block the PD-1/PD-L1 pathway.19,27 Historically, immunotherapy has been ineffective in cases of non–small-cell lung cancer (NSCLC), which has been thought to be a type of nonimmunogenic cancer; nevertheless, lung cancer can evade the immune system through various complex mechanisms.28 In patients with advanced lung cancer, the peripheral and tumor lymphocyte counts are decreased, while levels of regulatory T cells (CD4+), which help suppress tumor immune surveillance, have been found at higher levels.29-32 Immune checkpoint pathways involving the CTLA-4 or the PD-1/PD-L1 are involved in regulating T-cell responses, providing the rationale for blocking this pathway in NSCLC with antibodies against CTLA-4 and the PD-1/PD-L1 pathway.32 Triple negative breast cancer (TNBC) is an aggressive subset of breast cancer with limited treatment options. PD-L1 expression has been reported in patients with TNBC. When PD-L1 expression was evaluated in TILs, it correlated with higher grade and larger-sized tumors.33 Tumor PD-L1 expression also correlates with the infiltration of T-regulatory cells in TNBC, findings that suggest the role of PD-L1–expressing tumors and the PD-1/PD-L1–expressing TILs in regulating immune response in TNBC.34
…….
Preclinical evidence exists for the complementary roles of CTLA-4 and PD-1 in regulating adaptive immunity, and this provides rationale for combining drugs targeting these pathways.44-46 Paradoxically and originally believed to be immunosuppressive, new data allow us to recognize that cytotoxic agents can antagonize immunosuppression in the tumor microenvironment, thus promoting immunity based on the concept that tumor cells die in multiple ways and that some forms of apoptosis may lead to an enhanced immune response.8,15 For example, nivolumab was combined with ipilimumab in a phase 1 trial of patients with advanced melanoma.46 The combination had a manageable safety profile and produced clinical activity in the majority of patients, with rapid and deep tumor regression seen in a large proportion of patients. Based on the results of this study, a phase 3 study is being undertaken to evaluate whether this combination is better than nivolumab alone in melanoma (NCT01844505). Several other early-phase studies are underway to explore combinations of various anti–PD-1/PD-L1 drugs with other therapies across a variety of tumor types (see Tables 1 and 2), possibly paving the way for future combination studies.
Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer
Jiabei He, Ying Hu, Mingming Hu & Baolan Li
- Scientific Reports5, Article number: 13110 (2015) http://www.nature.com/articles/srep13110
Lung cancer is currently the leading cause of cancer-related death in worldwide, non-small cell lung cancer (NSCLC) accounts for about 85% of all lung cancers. Surgery, platinum-based chemotherapy, molecular targeted agents and radiotherapy are the main treatment of NSCLC. With the strategies of treatment constantly improving, the prognosis of NSCLC patients is not as good as before, new sort of treatments are needed to be exploited. Programmed death 1 (PD-1) and its ligand PD-L1 play a key role in tumor immune escape and the formation of tumor microenvironment, closely related with tumor generation and development. Blockading the PD-1/PD-L1 pathway could reverse the tumor microenvironment and enhance the endogenous antitumor immune responses. Utilizing the PD-1 and/or PD-L1 inhibitors has shown benefits in clinical trials of NSCLC. In this review, we discuss the basic principle of PD-1/PD-L1 pathway and its role in the tumorigenesis and development of NSCLC. The clinical development of PD-1/PD-L1 pathway inhibitors and the main problems in the present studies and the research direction in the future will also be discussed.
Lung cancer is currently the leading cause of cancer-related death in the worldwide. In China, the incidence and mortality of lung cancer is 5.357/10000, 4.557/10000 respectively, with nearly 600,000 new cases every year1. Non-small cell lung cancer (NSCLC) accounts for about 85% of all lung cancers, the early symptoms of patients with NSCLC are not very obvious, especially the peripheral lung cancer. Though the development of clinic diagnostic techniques, the majority of patients with NSCLC have been at advanced stage already as they are diagnosed. Surgery is the standard treatment in the early stages of NSCLC, for the advanced NSCLC, the first-line therapy is platinum-based chemotherapy. In recent years, patients with specific mutations may effectively be treated with molecular targeted agents initially. The prognosis of NSCLC patients is still not optimistic even though the projects of chemotherapy as well as radiotherapy are continuously ameliorating and the launch of new molecular targeted agents is never suspended, the five-year survival rate of NSCLC patients is barely more than 15%2, the new treatment is needed to be opened up.
During the last few decades, significant efforts of the interaction between immune system and immunotherapy to NSCLC have been acquired. Recent data have indicated that the lack of immunologic control is recognized as a hallmark of cancer currently. Programmed death-1 (PD-1) and its ligand PD-L1 play a key role in tumor immune escape and the formation of tumor microenvironment, closely related with tumor generation and development. Blockading the PD-1/PD-L1 pathway could reverse the tumor microenvironment and enhance the endogenous antitumor immune responses.
In this review, we will discuss the PD-1/PD-L1 pathway from the following aspects: the basic principle of PD-1/PD-L1 pathway and its role in the tumorigenesis and development of NSCLC, the clinical development of several anti-PD-1 and anti-PD-L1 drugs, including efficacy, toxicity, and application as single agent, or in combination with other therapies, the main problems in the present studies and the research direction in the future.
Cancer as a chronic, polygene and often inflammation-provoking disease, the mechanism of its emergence and progression is very complicated. There are many factors which impacted the development of the disease, such as: environmental factors, living habits, genetic mutations, dysfunction of the immune system and so on. At present, increasing evidence has revealed that the development and progression of tumor are accompanied by the formation of special tumor immune microenvironment. Tumor cells can escape the immune surveillance and disrupt immune checkpoint of host in several methods, therefore, to avoid the elimination from the host immune system. Human cancers contain a number of genetic and epigenetic changes, which can produce neoantigens that are potentially recognizable by the immune system3, thus trigger the body’s T cells immune response. The T cells of immune system recognize cancer cells as abnormal primarily, generate a population of cytotoxic T lymphocytes (CTLs) that can traffic to and infiltrate cancers wherever they reside, and specifically bind to and then kill cancer cells. Effective protective immunity against cancer depends on the coordination of CTLs4. Under normal physiological conditions, there is a balance status in the immune checkpoint molecule which makes the immune response of T cells keep a proper intensity and scope in order to minimize the damage to the surrounding normal tissue and avoid autoimmune reaction. However, numerous pathways are utilized by cancers to up-regulate the negative signals through cell surface molecules, thus inhibit T-cell activation or induce apoptosis and promote the progression and metastasis of cancers5. Increasing experiments and clinical trails show that immunotherapeutic approaches utilizing antagonistic antibodies to block checkpoint pathways, can release cancer inhibition and facilitate antitumor activity, so as to achieve the purpose of treating cancer.
The present research of immune checkpoint molecules are mainly focus on cytotoxic T lymphocyte-associated antigen 4 (CLTA-4), Programmed death-1 (PD-1) and its ligands PD-L1 (B7H1) and PD-L2 (B7-DC). CTLA-4 regulates T cell activity in the early stage predominantly, and PD-1 mainly limits the activity of T-cell in the tumor microenvironment at later stage of tumor growth6. Utilizing the immune checkpoint blockers to block the interactions between PD-1 and its ligands has shown benefits in clinical trials, including the NSCLC patients. PD-1 and its ligands have been rapidly established as the currently most important breakthrough targets in the development of effective immunotherapy.
PD-1/PD-L1 pathway and its expression, regulation
PD-1 is a type 1 trans-membrane protein that encoded by the PDCD1 gene7. It is a member of the extended CD28/CTLA-4 immunoglobulin family and one of the most important inhibitory co-receptors expressed by T cells. The structure of the PD-1 includes an extracellular IgV domain, a hydrophobic trans-membrane region and an intracellular domain. The intracellular tail includes separate potential phosphorylation sites that are located in the immune receptor tyrosine-based inhibitory motif (ITIM) and in the immunoreceptor tyrosine-based switch motif (ITSM). Mutagenetic researches indicated that the activated ITSM is essential for the PD-1 inhibitory effect on T cells8. PD-1 is expressed on T cells, B cells, monocytes, natural killer cells, dendritic cells and many tumor-infiltrating lymphocytes (TILs)9. In addition, the research of Francisoet et al. showed that PD-1 was also expressed on regulatory T cells (Treg) and able to facilitate the proliferation of Treg and restrain immune response10.
PD-1 has two ligands: PD-L1 (also named B7-H1; CD274) and PD-L2 (B7-DC; CD273), that are both coinhibitory. PD-L1 is expressed on resting T cells, B cells, dendritic cells, macrophage, vascular endothelial cells and pancreatic islet cells. PD-L2 expression is seen on macrophages and dendritic cells alone and is far less prevalent than PD-L1 across tumor types. It shows much more restricted expression because of its more restricted tissue distribution. Differences in expression patterns suggest distinct functions in immune regulation across distinct cell types. The restricted expression of PD-L2, largely to antigen-presenting cells, is consistent with a role in regulating T-cell priming or polarization, whereas broad distribution of PD-L1 suggests a more general role in protecting peripheral tissues from excessive inflammation.
PD-L1 is expressed in various types of cancers, especially in NSCLC11,12, melanoma, renal cell carcinoma, gastric cancer, hepatocellular as well as cutaneous and various leukemias, multiple myeloma and so on13,14,15. It is present in the cytoplasm and plasma membrane of cancer cells, but not all cancers or all cells within a cancer express PD-L116,17. The expression of PD-L1 is induced by multiple proinflammatory molecules, including types I and II IFN-γ, TNF-α, LPS, GM-CSF and VEGF, as well as the cytokines IL-10 and IL-4, with IFN-γ being the most potent inducer18,19. IFN-γ and TNF-α are produced by activated type 1 T cells, and GM-CSF and VEGF are produced by a variety of cancer stromal cells, the tumor microenvironment upregulates PD-L1 expression, thereby, promotes immune suppression. This latter effect is called “adaptive immune resistance”, because the tumor protects itself by inducing PD-L1 in response to IFN-γ produced by activated T cells17. PD-L1 is regulated by oncogenes, also known as the inherent immune resistance. PD-L1 expression is suppressed by the tumor suppressor gene: PTEN (phosphatase and tension homolog deleted on chromosome ten) gene. Cancer cells frequently contain mutated PTEN, which can activate the S6K1 gene, thus results in PD-L1 mRNA to polysomes increase greatly20, hence increases the translation of PD-L1 mRNA and plasma membrane expression of PD-L1. Parsa et al.’s research also demonstrated that neuroglioma with PTEN gene deletion regulate PD-L1 expression at the translational level by activating the PI3K/AKT downstream mTOR-S6K1signal pathway and, hence increase the PD-L1 expression21. Micro-RNAs also translationally regulate PD-L1 expression. MiRNA-513 is complementary to the 3′ untranslated region of PD-L1 and prevents PD-L1 mRNA translation22. In addition, a later literature reported that in the model of melanoma, the up-regulation of PD-L1 is closely related to the CD8 T cell, independent of regulation by oncogenes13. Noteworthily, the PD-L1 can bind to T cell expressed CD80, and at this point CD80 is a receptor instead of ligand to transmit negative regulated signals23.
PD-1/PD-L1 mediate immune suppression by multiple mechanisms
Like the CTLA-4, the PD-1/PD-L1 pathway down-modulates T-cell response by regulating overlapping signal proteins in the immune checkpoint pathway. However, their functions are slightly different24. The CTLA-4 focuses on regulating the activation of T cells, while PD-1 regulates effector T-cell activity in peripheral tissues in response to infection or tumor progression25. Tregs that high-level expression of PD-1 have been shown to have immune inhibitory activity, thus, they are important for maintaining self-tolerance. In normal human bodies, this is a crucial step to protect against tissue damage when the immune system is activated in response to infection26. However, in response to immune attack, cancer cells overexpress PD-L1 and PD-L2. They bind to PD-1 receptor on T cells, inhibiting the activation of T-cells, thus suppressing T-cell attack and inducing tumor immune escape. Thus tumor cells effectively form a suitable tumor microenvironment and continue to proliferate27. PD-1/PD-L1 pathway regulates immune suppression by multiple mechanisms, specific performance of the following: ① Induce apoptosis of activated T cells: PD-1 reduces T cell survival by impacting apoptotic genes. During T cell activation, CD28 ligation sustains T cell survival by driving expression of the antiapoptotic gene Bcl-xL. PD-1 prevents Bcl-xL expression by inhibiting PI3K activation, which is essential for upregulation of Bcl-xL. Early studies demonstrated that PD-L1+ murine and human tumor cells induce apoptosis of activated T cells and that antibody blocking of PD-L1 can decrease the apoptosis of T cells and facilitate antitumor immunity28,16. ② Facilitate T cell anergy and exhaustion: A research shown that the occurrence of tumor is associated with chronic infection29. According to the study of chronic infection, PD-1 overexpressed on the function exhausted T cells, blocking the PD-1/PD-L1 pathway can restore the proliferation, secretion and cytotoxicity30. In addition, later research demonstrated that the exhaustion of TILs in the tumor microenvironment is closely related to the PD-L1 expression of tumor cells, myeloid cells derived from tumor31. ③ Enhance the function of regulatory T cells: PD-L1 can promote the generation of induced Tregs by down-regulating the mTOR, AKT, S6 and the phosphorylation of ERK2 and increasing PTEN, thus restrain the activity of effector T-cell32. Blocking the PD-1/PD-L1 pathway can increase the function of effector CD8 T-cell and inhibt the function of Tregs, bone marrow derived inhibition cells, thus enhance the anti-tumor response. ④ Inhibit the proliferation of T cells: PD-1 ligation also prevents phosphorylation of PKC-theta, which is essential for IL-2 production33, and arrests T cells in the G1 phase, blocking proliferation. PD-1 mediates this effect by activating Smad3, a factor that arrests cycling34. ⑤ Restrain impaired T cell activation and IL-2 production: PD-1/PD-L1 blocks the downstream signaling events triggered by Ag/MHC engagement of the TCR and co-stimulation through CD28, resulting in impaired T cell activation and IL-2 production. Signaling through the TCR requires phosphorylation of the tyrosine kinase ZAP70. PD-1 engagement reduces the phosphorylation of ZAP70 and, hence, inhibits downstream signaling events. In addition, signaling through PD-1 also prevents the conversion of functional CD8+ T effector memory cells into CD8+ central memory cells35 and, thus, reduces long-term immune memory that might protect against future metastatic disease. PD-L1 also promotes tumor progression by reversing signaling through CD80 into T cells. CD80-PD-L1 interactions restrain self-reactive T cells in an autoimmune setting36, therefore, their inhibition may facilitate antitumor immunity.
Researches on the mechanism of PD-1/PD-L1 pathway mediating immune escape are still ongoing, especially the mechanism of PD-L2 is still unclear. These researches provide the theoretical basis and research direction for the further immunotherapy targets research.
Anti-PD-1 antibodies
Nivolumab
Nivolumab (BMS-936558, Brand name: Opdivo) is a human monoclonal IgG4 antibody that essentially lacks detectable antibody-dependent cellular cytotoxicity (ADCC). Inhibition by monoclonal antibody of PD-1 on CD8+ TILs within lung cancers can restore cytokine secretion and T-cell proliferation48. Results of a larger phase I study in 296 patients (236 patients evaluated) reported that the objective response (complete or partial responses) of patients with NSCLC was 18%. A total of 65% of responders had durable responses lasting for more than 1 year. Stable disease lasting 24 weeks was seen in patients with NSCLC. PD-L1expression was tested in 42 patients: 9 of 25(36%) patients whose PD-L1 expression positive were objectively response to PD-1 blockade treatment, while the remaining 17 nonresponsive patients were negative45.
In another early phase I trial of nivolumab49, an objective response was observed in 22 patients (17%; 95% CI, 11%–25%) in a dose-expansion cohort of 129 previously treated patients with advanced NSCLC. Six additional patients who had an unconventional immune-related response were not included. Moreover, the median duration of response was exceptional for 17 months. Although the median PFS in the cohort was 2.3 months and the median overall survival was 9.9 months, it seemed clear that those who responded had sustained benefit. Specifically, the 2-year overall survival rate was 24%, and many remained in remission after completing 96 weeks of continuous therapy.
Single-agent trials of nivolumab are planning or ongoing on NSCLC (NCT01721759, NCT02066636). In addition, there are clinical randomized trials which focus on the comparison of nivolumab and plain-based combination chemotherapy (NCT02041533, NCT01673867). In March 4, 2015, nivolumab was approved by the US Food and Drug Administration for treatment of patients with metastatic NSCLC (squamous cell carcinoma), when progression of their diseases during or after chemotherapy with platinum-based drugs.
Pembrolizumab
Pembrolizumab (MK-3475) is a highly selective, humanized monoclonal antibody with activity against PD-1 that contains a mutation at C228P designed to prevent Fc-mediated ADCC. It is now in the clinical research phases for patients with advanced solid tumors. Its safety and efficacy were evaluated in a phase I clinical trial of KEYNOTE-001. The best response according of 38 cases of patients which initially accepted pembrolizumab 10 mg/kg q3wwas 21% (based on RECIST1.1 evaluation) and the median PFS of responder still has not reached until 62 weeks. The researchers also found that the antitumor activity of pembrolizumab was associated with the PD-L1expression44,50. The critical values of the expression of PD-L1 will be validated in 300 cases of patients which will soon been rolled into the study.
Clinical trial of pembrolizumab monotherapy is ongoing for patients with NSCLC (NCT01840579). Randomized trials comparing pembrolizumab to combination chemotherapy (NCT02142738) or single-agent docetaxel (NCT01905657) are ongoing in PD-L1 positive patients with NSCLC.
Pidilizumab (CT-011)
Pidilizumab is a humanized IgG-1K recombinant anti-PD-1 monoclonal antibody that has demonstrated antitumor activity in mouse cancer models. In a first-in-human phase I dose-escalation study in patients with only advanced hematologic cancers, there is no clinical trials of NSCLC presently51.
Anti-PD-L1 antibodies
Another therapeutic method based on the PD-1/PD-L1 pathway is by specific binding between antibody and PD-L1, thus preventing its activity. It was speculated that utilizing PD-L1 as therapeutic target maybe accompanied by less toxicity in part by modulating the immune response selectively in the tumor microenvironment. However, since PD-L2 expressed by tumor cells or some other tumor-associated molecules may play a role in mediating PD-1-expressing lymphocytes, it is conceivable that the magnitude of the anti-tumor immune response could also be blunted.
BMS-936559
BMS-936559/MDX1105 is a fully humanized, high affinity, IgG4 monoclonal antibody that react specifically with PD-L1, thus inhibiting the binding of PD-L1 and PD-1, CD80 (which binds not only PD-L1 but also CTLA-4 and CD28). Initial results from a multicenter and dose-escalation phase I trial of 207 patients(including 75 cases of patients with NSCLC) showed durable tumor regression (objective response rate of 6%–17%) and prolonged stabilization of disease (12%–41% at 24weeks) in patients with advanced cancers, including NSCLC, melanoma and kidney cancer. In patients with NSCLC, there were five objective responses (in 4 patients with the nonsquamous subtype and 1 with the squamous subtype) at doses of 3 mg/kg and 10 mg/kg, with response rates of 8% and 16%, respectively. Six additional patients with NSCLC had stable disease lasting at least 24 weeks52.
MPDL3280A
MPDL3280A is a human IgG1 antibody that targets PD-L1. Its Fc component has been engineered to not activate antibody-dependent cell cytotoxicity. In a recently reported phase I study, a 21% response rate was noted in patients with metastatic melanoma, RCC or NSCLC53, including several patients who demonstrated shrinkage of tumor within a few days of initiating treatment.
Fifty-two patients were enrolled in an expansion cohort of the phase I trial of MPDL3280A, 62% of them were heavily pretreated NSCLC (≥3 lines of systemic therapy) and the ORR was 22%54. Analysis of biomarker data from archival tumor samples demonstrated a correlation between PD-L1 status and response and lack of progressive disease55.
MEDI4736
MEDI4736 is a human IgG1 antibody that binds specifically to PD-L1, thus preventing PD-L1 binding to PD-1 and CD80. Interim results from a phase I trial reported no colitis or pneumonitis of any grade, with several durable remissions, including NSCLC patients56. An ongoing phase I dose-escalation study (NCT01693562) of MEDI-4736 in 26 patients, 4 partial responses (3 in patients with NSCLC and 1 with melanoma) were observed and 5 additional patients exhibited lesser degrees of tumor shrinkage. The disease control rate at 12 weeks was 46%57. Expansion cohorts was opened in Sep 2013, 10 mg/kg q2w dose. 151 patients was enrolled so far in the expansion cohorts, tumor shrinkage was reported as early as the first assessment at 6 weeks and among the 13 patients with NSCLC, responses were sustained at 10 or more to 14.9 or more months58. In the NSCLC expansion cohort, the response rate was 16% in 58 evaluable patients and the disease control rate at 12 weeks was 35% with responses seen in all histologic subtypes as well as in a smaller proportion of PD-L1- tumors.
On the basis of the favorable toxicity profile and promising activity in a heavily pretreated NSCLC population, a global Phase III placebo controlled trial using the 10 mg/kg biweekly dose has been launched in Stage III patients who have not progressed following chemo-radiation (NCT02125461). The primary outcome measures are overall survival and progression-free survival.
AMP-224
AMP-224 was a B7-DC-Fc fusion protein which can block the PD-1 receptor competitively59. Some NSCLC patients were included in a first-in-man phase I trial of this fusion protein drug. A dose-dependent reduction in PD-1-high TILs was observed at 4 hours and 2 weeks after drug administration60.
A variety of approaches for combining PD-1/PD-L1 pathway inhibitors with other therapeutic methods have been explored over the past few years in an effort to offer more feasible therapeutic options for clinic to improve treatment outcomes. Approaches have included combinations with other immune checkpoint inhibitors, immunostimulatory cytokines (e.g. IFN-y) cytotoxic chemotherapy, platinum-based chemotherapy, radiotherapy, anti-angiogenic inhibitors, tumor vaccine and small-molecule molecularly targeted therapies many with promising results61,62. Studies indicated that PD-1/PD-L1 pathway inhibitors were most effective when combined with treatments that activating the immune system63.
Preclinical evidence exists for the complementary roles of CTLA-4 and PD-1 in regulating adaptive immunity, and this provides rationale for combining drugs targeting these pathways. In a Phase I study in 46 chemotherapy-naive patients with NSCLC, four cohorts of patients received ipilimumab (3 mg/kg) plus nivolumab for four cycles followed by nivolumab 3 mg/kg intravenously every 2 weeks. The ORR was 22% and did not correlate with PD-L1 status64.
In another Phase I study, 56 patients with advanced NSCLC were assigned based on histology to four cohorts to receive nivolumab (5–10 mg/kg) intravenously every 3 weeks plus one of four concurrent standard “platinum doublet” chemotherapy regimens. No dose de-escalation was required for dose-limiting toxicity. The ORR was 33–50% across arms and the 1-year OS rates were promising at 59–87%65.
…..
The research of cancer immunotherapy provides a new wide space for cancer treatment (including NSCLC), and compared with other therapeutic method, immunotherapy has its unique advantages, such as: relative safety, effectivity, less and low grade side effect and so on. Especially with the discovery and continued in-depth study of PD-1/PD-L1 pathway in the immune regulation mechanism, many significative conclusions were reported. Data from many clinical trails suggest that some patients with NSCLC have been benefited from the drugs of anti-PD-1 and anti-PD-L1 already. However, summarized what have been discussed above, only a small fraction of patients benefit from PD-1 or PD-L1 inhibitors treatment. But with the continuous studies on biomarker and combined treatment in PD-1/PD-L1 pathway, new research progress will be acquired as well. We will make significant progress on treatment and in control of NSCLC.
Prospects for Targeting PD-1 and PD-L1 in Various Tumor Types
![]()
Review Article | November 10, 2014 | Oncology Journal, Supplements
 Table 1: Selected Anti–PD-1 and Anti–PD-L1 Antibodies
Table 1: Selected Anti–PD-1 and Anti–PD-L1 Antibodies Table 2: Selected Adverse Events
Table 2: Selected Adverse Events Table 3: Selected Clinical Trials for Metastatic Melanoma
Table 3: Selected Clinical Trials for Metastatic Melanoma Table: 4 Selected Trials for Metastatic Renal Cell Carcinoma
Table: 4 Selected Trials for Metastatic Renal Cell Carcinoma") Table 5: Selected Trials for Non–Small-Cell Lung Cancer (NSCLC )
Table 5: Selected Trials for Non–Small-Cell Lung Cancer (NSCLC ) Table 6: Selected Trials for Other Tumor Types
Table 6: Selected Trials for Other Tumor TypesImmune checkpoints, such as programmed death ligand 1 (PD-L1) or its receptor, programmed death 1 (PD-1), appear to be Achilles’ heels for multiple tumor types. PD-L1 not only provides immune escape for tumor cells but also turns on the apoptosis switch on activated T cells. Therapies that block this interaction have demonstrated promising clinical activity in several tumor types. In this review, we will discuss the current status of several anti–PD-1 and anti–PD-L1 antibodies in clinical development and their direction for the future.
Several PD-1 and PD-L1 antibodies are in clinical development (Table 1). Overall, they are very well tolerated; most did not reach dose-limiting toxicity in their phase I studies. As listed in Table 2, no clinically significant difference in adverse event profiles has been seen between anti–PD-1 and anti–PD-L1 antibodies. Slightly higher rates of infusion reactions (11%) were observed with BMS-936559 (anti–PD-L1) than with BMS-96558 (nivolumab). In an early stage of a nivolumab phase I study, there was concern about fatal pneumonitis.[7] It has been hypothesized that PD-1 interaction with PD-L2 expressed on the normal parenchymal cells of lung and kidney provides unique negative signaling that prevents autoimmunity.[8] Thus, anti–PD-1 antibody blockage of such an interaction may remove this inhibition, allowing autoimmune pneumonitis or nephritis. Anti–PD-L1 antibody, however, would theoretically leave PD-1–PD-L2 interaction intact, preventing the autoimmunity caused by PD-L2 blockade. With implementation of an algorithm to detect early signs of pneumonitis and other immune-related adverse events, many of these side effects have become manageable. However, it does require discerning clinical attention to detect potentially fatal side effects. In terms of antitumor activity, both anti–PD-1 and anti–PD-L1 antibodies have shown responses in overlapping multiple tumor types. Although limited to a fraction of patients, most responses, when observed, were rapid and durable.
– See more at: http://www.cancernetwork.com/oncology-journal/prospects-targeting-pd-1-and-pd-l1-various-tumor-types#sthash.an8uOYLi.dpuf
Immune Checkpoint Blockade in Cancer Therapy
Michael A. Postow, Margaret K. Callahan and Jedd D. Wolchok
http://jco.ascopubs.org/content/early/2015/01/20/JCO.2014.59.4358.full http://dx.doi.org:/10.1200/JCO.2014.59.4358
Immunologic checkpoint blockade with antibodies that target cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) and the programmed cell death protein 1 pathway (PD-1/PD-L1) have demonstrated promise in a variety of malignancies. Ipilimumab (CTLA-4) and pembrolizumab (PD-1) are approved by the US Food and Drug Administration for the treatment of advanced melanoma, and additional regulatory approvals are expected across the oncologic spectrum for a variety of other agents that target these pathways. Treatment with both CTLA-4 and PD-1/PD-L1 blockade is associated with a unique pattern of adverse events called immune-related adverse events, and occasionally, unusual kinetics of tumor response are seen. Combination approaches involving CTLA-4 and PD-1/PD-L1 blockade are being investigated to determine whether they enhance the efficacy of either approach alone. Principles learned during the development of CTLA-4 and PD-1/PD-L1 approaches will likely be used as new immunologic checkpoint blocking antibodies begin clinical investigation.
CTLA-4 was the first immune checkpoint receptor to be clinically targeted (Fig 1) Normally, after T-cell activation, CTLA-4 is upregulated on the plasma membrane where it functions to downregulate T-cell function through a variety of mechanisms, including preventing costimulation by outcompeting CD28 for its ligand, B7, and also by inducing T-cell cycle arrest.1–5 Through these mechanisms and others, CTLA-4 has an essential role in maintaining normal immunologic homeostasis, as evidenced by the fact that mice deficient in CTLA-4 die from fatal lymphoproliferation.6,7 Recognizing the role of CTLA-4 as a negative regulator of immunity, investigators led studies demonstrating that antibody blockade of CTLA-4 could result in antitumor immunity in preclinical models.8,9


IMAGE SOURCE:
http://ascopubs.org/doi/figure/10.1200/JCO.2014.59.4358
Article SOURCE:
http://jco.ascopubs.org/content/early/2015/01/20/JCO.2014.59.4358/F1.medium.gif
{kind=link}
Fig 1.
The cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) immunologic checkpoint. T-cell activation requires antigen presentation in the context of a major histocompatibility complex (MHC) molecule in addition to the costimulatory signal achieved when B7 on an antigen-presenting cell (dendritic cell shown) interacts with CD28 on a T cell. Early after activation, to maintain immunologic homeostasis, CTLA-4 is translocated to the plasma membrane where it downregulates the function of T cells.
On the basis of this preclinical rationale, two antibodies targeting CTLA-4, ipilimumab (Bristol-Myers Squibb, Princeton, NJ) and tremelimumab (formerly Pfizer, currently MedImmune/AstraZeneca, Wilmington, DE), entered clinical development. Early reports of both agents showed durable clinical responses in some patients.10–12Unfortunately, despite a proportion of patients experiencing a durable response, tremelimumab did not statistically significantly improve overall survival, which led to a negative phase III study comparing tremelimumab to dacarbazine/temozolomide in patients with advanced melanoma.13 It is possible that the lack of an overall survival benefit was a result of the crossover of patients treated with chemotherapy to an expanded access ipilimumab program or a result of the dosing or scheduling considerations of tremelimumab.
Ipilimumab, however, was successful in improving overall survival in two phase III studies involving patients with advanced melanoma.14,15 Although the median overall survival was only improved by several months in each of these studies, landmark survival after treatment initiation favored ipilimumab; in the first phase III study, 18% of patients were alive after 2 years compared with 5% of patients who received the control treatment of gp100 vaccination.14 More recently reported pooled data from clinical trials of ipilimumab confirm that approximately 20% of patients will have long-term survival of at least 3 years after ipilimumab therapy, with the longest reported survival reaching 10 years.16–18
For patients with other malignancies, CTLA-4 antibody therapy has also shown some benefits. Ipilimumab, in combination with carboplatin and paclitaxel in a phased treatment schedule, showed improved progression-free survival compared with carboplatin and paclitaxel alone for patients with non–small-cell lung cancer.19Several patients with pancreatic cancer had declines in CA 19-9 when ipilimumab was given with GVAX (Aduro, Berkeley, CA),20and ipilimumab has also resulted in responses in patients with prostate cancer.21 Unfortunately, a phase III study in patients with castrate-resistant prostate cancer who experienced progression on docetaxel chemotherapy demonstrated that after radiotherapy, ipilimumab did not improve overall survival compared with placebo.22 Although this study is felt to have been a negative study, ipilimumab may have conferred a benefit to patients with favorable prognostic features, such as the absence of visceral metastases, but this requires further study. Another CTLA-4–blocking antibody, tremelimumab, has shown responses in patients with mesothelioma, and ongoing trials are under way.23
CTLA-4 blockade has also been administered together with other immunologic agents, such as the indoleamine 2,3-dioxygenase inhibitor INCB024360,106 the oncolytic virus talimogene laherparepvec,107 and granulocyte-macrophage colony-stimulating factor,108 with encouraging early results. We expect subsequent studies involving engineered T-cell–based therapies and checkpoint blockade.
Other promising data involve CTLA-4 combinations with PD-1 blockade. A phase I study of ipilimumab and nivolumab in patients with melanoma resulted in a high durable response rate and impressive overall survival compared with historical data.109,110Although the most recently reported grade 3 or 4 toxicity rate in patients with melanoma was 64%, which is higher than either ipilimumab or nivolumab individually,111 the vast majority of these irAEs were asymptomatic laboratory abnormalities of unclear clinical consequence. For example, elevations in amylase or lipase were reported in 21% of patients, none of whom met clinical criteria for a diagnosis of pancreatitis. The rate of grade 3 or 4 diarrhea was 7%, which is approximately similar to the rate of grade 3 or 4 diarrhea with ipilimumab monotherapy at 3 mg/kg. Whether ipilimumab and nivolumab improve overall survival compared with either nivolumab or ipilimumab alone remains the subject of an ongoing phase III randomized trial, and investigations of the combination of ipilimumab and nivolumab (and tremelimumab and MEDI4736) are ongoing in many other cancers.
Immunotherapy with checkpoint-blocking antibodies targeting CTLA-4 and PD-1/PD-L1 has improved the outlook for patients with a variety of malignancies. Despite the promise of this approach, many questions remain, such as the optimal management of irAEs and how best to evaluate combination approaches to determine whether they will increase the efficacy of CTLA-4 or PD-1/PD-L1 blockade alone. Themes from the experience with CTLA-4 and PD-1/PD-L1 will likely be relevant for investigations of novel immunologic checkpoints in the future.
This is a very important article, Dr. Larry.
It fits so beautiful with our work on Molecules in Development Table.
Thank you

{kind=link}
This image depicts the process of metastasis in a mouse tumor, where tumor cells (green) have helped to reorganize the collagen into aligned fibers (blue) that provide the structural support for motility. This helps the tumor cells to enter blood vessels (red), ultimately leading to the formation of metastases in other organs. Image: Madeleine Oudin and Jeff Wyckoff
Paving the way for metastasis
Cancer cells remodel their environment to make it easier to reach nearby blood vessels.
Anne Trafton | MIT News Office March 15, 2016
A new study from MIT reveals how cancer cells take some of their first steps away from their original tumor sites. This spread, known as metastasis, is responsible for 90 percent of cancer deaths.
Studying mice, the researchers found that cancer cells with a particular version of the Mena protein, called MenaINV (invasive), are able to remodel their environment to make it easier for them to migrate into blood vessels and spread through the body. They also showed that high levels of this protein are correlated with metastasis and earlier deaths among breast cancer patients.
Finding a way to block this protein could help to prevent metastasis, says Frank Gertler, an MIT professor of biology and a member of the Koch Institute for Integrative Cancer Research.
“That’s something that I think would be very promising, because we know that when we genetically remove MenaINV, the tumors become nonmetastatic,” says Gertler, who is the senior author of a paper describing the findings in the journal Cancer Discovery.
Madeleine Oudin, a postdoc at the Koch Institute, is the paper’s lead author.
On the move
For cancer cells to metastasize, they must first become mobile and then crawl through the surrounding tissue to reach a blood vessel. In the new study, the MIT team found that cancer cells follow the trail of fibronectin, a protein that is part of the “extracellular matrix” that provides support for surrounding cells. Fibronectin is found in particularly high concentrations around the edges of tumors and near blood vessels.
“Cancer cells within a tumor environment are constantly faced with differences in fibronectin concentrations, and they need to be able to move from low to high concentrations to reach the blood vessels,” Oudin says.
MenaINV, an alternative form of the normal Mena protein, is key to this process. MenaINV includes a segment not found in the normal version, and this makes it bind more strongly to a receptor known as alpha-5 integrin, which is found on the surfaces of tumor cells and nearby supporting cells, and recognizes fibronectin.
When MenaINV attaches to this receptor, it promotes the binding of fibronectin to the same receptors. Fibronectin is normally a tangled protein, but when it binds to cell surfaces, it gets stretched out into long bundles. This stimulates the organization of collagen, another extracellular matrix protein, into stiff fibrils that radiate from the edges of the tumor.
This pattern, which is typically seen in tumors that are more aggressive, essentially paves the way for tumor cells to move toward blood vessels.
“If you have curly, coiled collagen, that’s associated with a good outcome, but if it gets realigned into these really straight long fibers, that provides highways for these cells to migrate on,” Oudin says.
In studies of mice, cells with the invasive form of Mena were better able to recognize and crawl toward higher concentrations of fibronectin, moving along the collagen pathways, while cells without MenaINV did not move toward the higher concentrations.
Predicting metastasis
The researchers also looked at data from breast cancer patients and found that high levels of MenaINV and fibronectin are associated with metastasis and earlier death. However, there was no link between the normal version of Mena and earlier death.
Gertler’s lab had previously developed antibodies that can detect the normal and invasive forms of Mena, which are now being developed for testing patient biopsy samples. Such tests could help doctors to determine whether a patient’s tumor is likely to spread or not, and possibly to guide the patient’s treatment. In addition, scientists may be able to develop drugs that inhibit MenaINV, which could be useful for treating cancer or preventing it from metastasizing.
The researchers now hope to study how MenaINV may contribute to other types of cancers. Preliminary studies suggest that it plays a similar role in lung and colon cancers as that seen in breast cancer. They are also investigating how the choice between the two forms of the Mena protein is regulated, and how other proteins found in the extracellular matrix might contribute to cancer cell migration.
Facilitating Tumor Cell Migration
Researchers identify a modified form of a migration-regulating protein in cancer cells that remodels the tumor microenvironment to promote metastasis.
| March 16, 2016
Emerging evidence suggests that metastasis—the spread of cancer from one organ or tissue to another—is aided by a significant remodeling of the cancer cells’ surroundings. Now, researchers at MIT have made progress toward understanding the mechanisms involved in this process by highlighting the role of a protein that reorganizes the tumor’s extracellular matrix to facilitate cellular migration into blood vessels. The findings were published yesterday (March 15) in Cancer Discovery.
Using a mouse model, the team showed that a cancer-cell-expressed protein called MenaINV—a mutated, “invasive” form of the cell-migration-modulator Mena—binds more strongly than its normal equivalent to a receptor on tumor and nearby support cells. The binding rearranges fibronectin in the tumor microenvironment, which in turn triggers the reorganization of collagen in the extracellular matrix into linear fibers radiating from the tumor.
This collagen restructuring is key in facilitating the migration of tumor cells to the blood vessels, from where they can disseminate throughout the body.
Tumor cell-driven extracellular matrix remodeling enables haptotaxis during metastatic progression
Madeleine J. Oudin1, Oliver Jonas1, Tatsiana Kosciuk1, Liliane C. Broye1, Bruna C. Guido1, Jeff Wyckoff1, …., James E. Bear2 and Frank B. Gertler1,*
Cancer Discov CD-15-1183 Jan 25, 2016 http://dx.doi.org:/10.1158/2159-8290.CD-15-1183
Fibronectin (FN) is a major component of the tumor microenvironment, but its role in promoting metastasis is incompletely understood. Here we show that FN gradients elicit directional movement of breast cancer cells, in vitro and in vivo. Haptotaxis on FN gradients requires direct interaction between α5β1 integrin and Mena, an actin regulator, and involves increases in focal complex signaling and tumor-cell-mediated extracellular matrix (ECM) remodeling. Compared to Mena, higher levels of the pro-metastatic MenaINV isoform associate with α5, which enables 3D haptotaxis of tumor cells towards the high FN concentrations typically present in perivascular space and in the periphery of breast tumor tissue. MenaINV and FN levels were correlated in two breast cancer cohorts, and high levels of MenaINV were significantly associated with increased tumor recurrence as well as decreased patient survival. Our results identify a novel tumor-cell-intrinsic mechanism that promotes metastasis through ECM remodeling and ECM guided directional migration.
Researchers Find Link Between Death of Tumor-support Cells and Cancer Metastasis Fri, 02/19/2016
http://www.dddmag.com/news/2016/02/researchers-find-link-between-death-tumor-support-cells-and-cancer-metastasis#.VuatbTol_kI.linkedin

http://www.dddmag.com/sites/dddmag.com/files/20160219-metastasized-cells%20%281%29.jpg
{kind=link}
The images show tumors that have metastasized to the lungs (image b) and bones (image d) in mice that had CAFs eliminated after 10 days. (Credit: Biju Parekkadan, Massachusetts General Hospital)
Researchers have discovered that eliminating cells thought to aid tumor growth did not slow or halt the growth of cancer tumors. In fact, when the cancer-associated fibroblasts (CAFs), were eliminated after 10 days, the risk of metastasis of the primary tumor to the lungs and bones of mice increased dramatically. Scientists used bioengineered CAFs equipped with genes that caused those cells to self-destruct at defined moments in tumor progression. The study, published in Scientific Reports on Feb. 19, was conducted by researchers funded by the National Institute of Biomedical Imaging and Bioengineering (NIBIB) at Massachusetts General Hospital (MGH). NIBIB is part of the National Institutes of Health.
What causes cancer to grow and metastasize is not well understood by scientists. CAFs are thought to be fibroblast cells native to the body that cancer cells hijacks and use to sustain their growth. However, because fibroblasts are found throughout the human body, it can be difficult to follow and study cancer effects on these cells.
“This work underscores two important things in solving the puzzle that is cancer,” said Rosemarie Hunziker, Ph.D., program director for Tissue Engineering at NIBIB. “First, we are dealing with a complex disease with so many dimensions that we are really only just beginning to describe it. Second, this approach shows the power of cell engineering — manipulating a key cell in the cancer environment has led to a significant new understanding of how cancer grows and how it might be controlled in the future.”
Biju Parekkadan, Ph.D., assistant professor of surgery and bioengineering at MGH, and his team designed an experiment with the goal of better understanding the cellular environment in which tumors exist (called tumor microenvironment or TME), and the role of CAFs in tumor growth. In an effort to understand whether targeting CAFs could limit the growth of breast cancer tumors implanted in mice, they bioengineered CAFs with a genetic “kill switch.” The cells were designed to die when exposed to a compound that was not toxic to the surrounding cells.
Parekkadan and his team chose two different stages of tumor growth in which the CAFs were killed off after the tumor was implanted. When the CAFs were eliminated on the third or fourth day, they found no major difference in tumor growth or risk of metastasis compared with the tumors where the CAFs remained. However, there was an increase in tumor-associated macrophages — cells that have been associated with metastasis — in this early stage.
When the team waited to eliminate the CAFs until the 10th or 11th day, they discovered that in addition to the increase in macrophages, the cancer was more likely to spread to the lungs and bones of the mice. The unexpected results from this experiment could spur more research into the role of CAFs in cancer growth and metastasis.
More research may reveal whether or not there is a scientific basis for targeting CAFs for destruction — and if so, the awareness that timing matters when it comes to the response of the tumor. While neither treatment affected the growth of the initial tumor, it is important to understand that most cancer deaths result from metastases to vital organs rather than from the direct effects of the primary tumor.