Long Term Memory and Prions
Larry H. Bernstein, MD, FCAP, Curator
LPBI
updated 12/12/2015
Possible biochemical mechanism underlying long-term memories identified

It’s a nagging question: why do some of our memories fade away, while others last forever? Now scientists at the Stowers Institute for Medical Research have identified a possible biochemical mechanism: a specific synaptic protein called Orb2 can either block or maintain neural synapses (connections between neurons), which create and maintain long-term memories.
So for a memory to persist, the synaptic connections must be kept strong. But how? The researchers previously identified a synaptic protein called CPEB that is responsible for maintaining the strength of such connections in the sea slug (a model organism used in memory research). Recently, they identified a similar protein, called Orb2, in the fruit fly.
Now, using a fruit fly model system, they found that the synaptic connections are kept strong by the transformation of Orb2 from one molecular state to another. And that transformation causes Orb2 molecules to solidify and strengthen the memory connections in the brain.
The authors conclude their paper, published in the current issue of the journal Cell, with several questions. How and what triggers this transformation, how long does it persist? Is the continued presence of a prion-like state necessary for the persistence of memory, and is it correlated with or predictive of long-lasting memory? And most interestingly: can a transient memory about to be forgotten be stabilized by artificial recruitment of the prion-like state (perhaps by a neurotropic compound)?
And what about that ironic link with prions, associated with neurodegenerative disorders? Are prions some twisted form of memory that could one day even have value? We’ll be keeping an eye on where this fascinating research leads.
Technical details: the memory switch
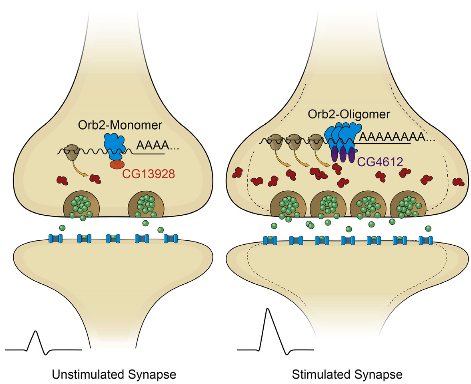
In their latest study, the researchers determined that Orb2 exists in two distinct physical states: monomeric (a single molecule that can bind to other molecules) and oligomeric (a molecular complex).
Like CPEB, oligomeric Orb2 is prion-like — that is, it’s a self-copying cluster. (But unlike prions, oligomeric Orb2 and CPEB are not toxic.) Monomeric Orb2 represses, and oligomeric Orb2 activates a crucial step in the complex cellular process that leads to protein synthesis.
During this crucial step, messenger RNA (mRNA), which is an RNA copy of a gene’s recipe for a protein, is translated by the cell’s ribosome into the sequence of amino acids that will make up a newly synthesized protein. The monomeric form of Orb2 binds to the target mRNA, keeping it in a repressed state.
The Stowers scientists also determined that prion-like Orb2 not only activates translation into amino acids but imparts its translational state to nearby monomer forms of Orb2. As a result, monomeric Orb2 is transformed into prion-like Orb2, so its role in translation switches from repression to activation.
Self-sustaining activation maintains synaptic activity
Stowers Associate Investigator Kausik Si, Ph.D. thinks this switch is the possible mechanism by which fleeting experiences create an enduring memory. “Because of the self-sustaining nature of the prion-like state, this creates a local and self-sustaining translation activation of Orb2-target mRNA, which maintains the changed state of synaptic activity over time,” says Si.
The discovery that the two distinct states of Orb2 have opposing roles in the translation process provides “for the first time a biochemical mechanism of synapse-specific persistent translation and long-lasting memory,” he states.
“To our knowledge, this is the first example of a prion-based protein switch that turns a repressor into an activator,” Si adds. “The recruitment of distinct protein complexes at the non-prion and prion-like forms to create altered activity states indicates the prion-like behavior is in essence a protein conformation-based switch.
“Through this switch, a protein can lose or gain a function that can be maintained over time in the absence of the original stimuli. Although such a possibility has been anticipated prior to this study, there was no direct evidence.”
The research builds upon previous studies by Si and Eric Kandel, M.D., of Columbia University and other scientists. These studies revealed that both short-term and long-term memories are created in synapses.
Abstract of Amyloidogenic Oligomerization Transforms Drosophila Orb2 from a Translation Repressor to an Activator
Memories are thought to be formed in response to transient experiences, in part through changes in local protein synthesis at synapses. In Drosophila, the amyloidogenic (prion-like) state of the RNA binding protein Orb2 has been implicated in long-term memory, but how conformational conversion of Orb2 promotes memory formation is unclear. Combining in vitro and in vivo studies, we find that the monomeric form of Orb2 represses translation and removes mRNA poly(A) tails, while the oligomeric form enhances translation and elongates the poly(A) tails and imparts its translational state to the monomer. The CG13928 protein, which binds only to monomeric Orb2, promotes deadenylation, whereas the putative poly(A) binding protein CG4612 promotes oligomeric Orb2-dependent translation. Our data support a model in which monomeric Orb2 keeps target mRNA in a translationally dormant state and experience-dependent conversion to the amyloidogenic state activates translation, resulting in persistent alteration of synaptic activity and stabilization of memory.
references:
Cell 2015 ; http://dx.doi.org:/10.1016/j.cell.2015.11.020
New Finding on Synapse Destruction May Open Path to Alzheimer’s Therapy
A team led by scientists at the University of New South Wales in Australia say they have discovered how connections between brain cells are destroyed in the early stages of Alzheimer’s disease. They believe their work opens up a new avenue for research on possible treatments for the degenerative brain condition.
“One of the first signs of Alzheimer’s disease is the loss of synapses—the structures that connect neurons in the brain,” noted study leader, Vladimir Sytnyk, Ph.D., of the UNSW School of Biotechnology and Biomolecular Sciences. “Synapses are required for all brain functions, and particularly for learning and forming memories. In Alzheimer’s disease, this loss of synapses occurs very early on, when people still only have mild cognitive impairment, and long before the nerve cells themselves die. We have identified a new molecular mechanism which directly contributes to this synapse loss, a discovery we hope could eventually lead to earlier diagnosis of the disease and new treatments.”
The team studied a specific protein in the brain, neural cell adhesion molecule 2 (NCAM2), one of a family of molecules that physically connects the membranes of synapses and help stabilize these long lasting synaptic contacts between neurons. The researchers paper (“Aβ-dependent reduction of NCAM2-mediated synaptic adhesion contributes to synapse loss in Alzheimer’s disease”) is published in Nature Communications.
Using post-mortem brain tissue from people with and without the condition, they discovered that synaptic NCAM2 levels in the part of the brain known as the hippocampus were low in those with Alzheimer’s disease. They also showed in mice studies and in the laboratory that NCAM2 was broken down by beta-amyloid, which is the main component of the plaques that build up in the brains of people with the disease.
“Our research shows the loss of synapses is linked to the loss of NCAM2 as a result of the toxic effects of beta-amyloid,” pointed out Dr. Sytnyk. “It opens up a new avenue for research on possible treatments that can prevent the destruction of NCAM2 in the brain.”
Aβ-dependent reduction of NCAM2-mediated synaptic adhesion contributes to synapse loss in Alzheimer’s disease
Iryna Leshchyns’ka, Heng Tai Liew, Claire Shepherd, Glenda M. Halliday, Claire H. Stevens, Yazi D. Ke, Lars M. Ittner & Vladimir Sytnyk
Nature Communications Nov 2015; 6(8836) doi:10.1038/ncomms9836
Alzheimer’s disease (AD) is characterized by synapse loss due to mechanisms that remain poorly understood. We show that the neural cell adhesion molecule 2 (NCAM2) is enriched in synapses in the human hippocampus. This enrichment is abolished in the hippocampus of AD patients and in brains of mice overexpressing the human amyloid-β (Aβ) precursor protein carrying the pathogenic Swedish mutation. Aβ binds to NCAM2 at the cell surface of cultured hippocampal neurons and induces removal of NCAM2 from synapses. In AD hippocampus, cleavage of the membrane proximal external region of NCAM2 is increased and soluble extracellular fragments of NCAM2 (NCAM2-ED) accumulate. Knockdown of NCAM2 expression or incubation with NCAM2-ED induces disassembly of GluR1-containing glutamatergic synapses in cultured hippocampal neurons. Aβ-dependent disassembly of GluR1-containing synapses is inhibited in neurons overexpressing a cleavage-resistant mutant of NCAM2. Our data indicate that Aβ-dependent disruption of NCAM2 functions in AD hippocampus contributes to synapse loss.
Learning and memory processes depend on the number and correct functioning of synapses in the brain. Cell adhesion molecules are enriched in the pre- and postsynaptic membranes. These molecules physically connect synaptic membranes, providing mechanical stabilization of synaptic contacts1, 2, 3, are necessary for the formation of new synapses during neuronal development4, 5, and maintain and regulate synaptic plasticity in adults6, 7, 8, 9, 10.
Alzheimer’s disease (AD) is a neurodegenerative brain condition predominantly of the aging population. One of the earliest signs of AD is the loss of synapses11, which can at least partially be linked to the toxicity mediated by Aβ12, 13, 14, a peptide that accumulates in the brains of AD patients. The impact of AD on synaptic adhesion and the role of synaptic cell adhesion molecules in the progression of the disease remains poorly understood.
The neural cell adhesion molecule 2 (NCAM2), sometimes designated OCAM, belongs to the immunoglobulin superfamily of cell adhesion molecules. NCAM2 participates in homophilic trans-interactions15, 16. During human embryonic development, NCAM2 is expressed in several tissues, including lung, liver, and kidney with the highest expression in the brain17. The expression level of NCAM2 peaks around postnatal day 21 and remains high during adulthood15, suggesting that the protein is necessary both during development and in adult brains. Accordingly, studies with cultured neurons and in NCAM2 deficient mice show that NCAM2 is important for the development of the brain, and the olfactory system in particular18, 19.
The NCAM2 gene is located on chromosome 21 in humans and NCAM2 overexpression has been suggested to be one of the factors contributing to the symptoms of Down syndrome17, which presents with early-onset AD pathology. Single-nucleotide polymorphisms in the NCAM2 gene have been reported as a risk factor related to the progression of AD in the Japanese population20. A recent genome-wide association study has found an association between single-nucleotide polymorphisms in the NCAM2 gene and levels of Aβ in the cerebrospinal fluid in humans, suggesting that NCAM2 is involved in the pathogenic pathway to the senile plaques that concentrate in AD brains21. Since genetic association studies indicate a link between NCAM2 and AD, we have analysed whether AD pathology influences levels of NCAM2 in synapses. Our results indicate that the synaptic adhesion mediated by NCAM2 is highly susceptible to Aβ toxicity and that proteolytic fragments of NCAM2 generated in an Aβ-dependent manner can directly contribute to the induction of synapse disassembly.
Synaptic NCAM2 is reduced in the hippocampus in AD
To analyse whether functions of NCAM2 are affected in AD, frozen post-mortem brain tissue of AD patients and non-affected controls (n=10 each) was analysed by western blot with antibodies against NCAM2. The detailed demographic data for the subjects analysed are presented inSupplementary Table 1. Total levels of NCAM2 were slightly increased in the hippocampus, but not significantly affected in the cerebellum or superior temporal cortex in AD (Supplementary Fig. 1). In contrast, levels of VGLUT1, a presynaptic marker-protein of excitatory synapses, were reduced in AD hippocampus (Supplementary Fig. 1), indicating a loss of excitatory synapses. Levels of VGAT, a presynaptic marker-protein of inhibitory synapses, were not significantly affected in any brain region analysed (Supplementary Fig. 1).
Changes in the protein levels in brain homogenates do not necessarily reflect changes in the protein levels in synapses. To analyse whether the synaptic function of NCAM2 is affected in AD, we compared the enrichment of NCAM2 in synaptosomes isolated from the brain tissue of individuals with AD and non-affected controls by western blot analysis of synaptosomes and total homogenates of the brains used for synaptosome preparations. Equal total protein amounts from each probe were applied to the gels to compensate for any possible differences in the yield of synaptosomes because of the synapse loss observed in AD. Western blot analysis with antibodies against actin, VGLUT1, VGAT, synaptophysin (a general presynaptic marker-protein), and PSD95 (a postsynaptic marker-protein), showed that these proteins were enriched to similar levels in synaptosomes from AD and control brains, indicating similar purities of intact synaptosome isolations (Fig. 1a). Western blot analysis showed that in control individuals NCAM2 was highly enriched in synaptosomes from the hippocampus and to a lower degree in synaptosomes from the temporal cortex and cerebellum (Fig. 1a,b). This synaptic enrichment of NCAM2 was significantly reduced in synaptosomes from AD hippocampi (Fig. 1a,b). The synaptic enrichment of NCAM2 was slightly lower in the AD versus control cerebellum, however the difference was not statistically significant (Fig. 1a,b).
Figure 1: Synaptic accumulation of NCAM2 is reduced in the hippocampus of AD-affected individuals.
Figure 2: Cleavage of the membrane-adjacent extracellular fragment of NCAM2 is increased in AD brains.
Figure 3: The extracellular domain of NCAM2 binds to Aβ.
Cleavage of NCAM2aa682-701 is increased in AD brains
NCAM2 binds to Aβ in vitro
Figure 4: NCAM2 accumulates in excitatory synapses of cultured hippocampal neurons.

http://www.nature.com/ncomms/2015/151127/ncomms9836/images_article/ncomms9836-f4.jpg
(a) Low-magnification image of a cultured hippocampal neuron labelled by indirect immunofluorescence with antibodies against NCAM2, synaptophysin and MAP2. Note expression of NCAM2 along MAP2 positive dendrites. NCAM2 is also expressed in astrocytes (marked a) which are present in these cultures. Scale bar, 20 μm. (b) High-magnification image of dendrites of neurons co-labelled with antibodies against NCAM2, synaptophysin and MAP2. Arrows show clusters of NCAM2 partially overlapping with synaptophysin accumulations. NCAM2-negative synapses are also observed (arrowheads). Scale bar, 10 μm. (c) High-magnification image of a dendrite of a cultured hippocampal neuron labelled with antibodies against NCAM2, synaptophysin and PSD95. NCAM2 clusters partially overlap with accumulations of PSD95 and synaptophysin (arrows). Scale bar, 10 μm. Three-dimensional analysis of the co-localization within the outlined area is on the right. Z-stack has been acquired with 0.15 μm steps. The xz and yz sections along the dashed lines on the xy image are shown. Note co-localization of the NCAM2 cluster with synaptic markers. (d) Negative control, that is, labelling performed without primary antibodies, is shown. Scale bar, 10 μm.
Figure 5: Aβ1–42 oligomers bind to NCAM2 at the cell surface of neurons.
Figure 6: Levels of NCAM2 are reduced in synaptosomes of cultured hippocampal neurons treated with Aβ1-42 oligomers.
Figure 7: NCAM2 co-localizes with Aβ1-42 in brains of APP23 transgenic mice.
Figure 8: NCAM2 binds to Aβ and its synaptic accumulation is reduced in the hippocampus of APP23 transgenic mice.
Aβ removes NCAM2 from synapses of hippocampal neurons
Western blot analysis showed that levels of soluble NCAM2 with the molecular weight of ~100 kDa were significantly increased in culture medium from Aβ1-42-treated hippocampal neurons (Fig. 6b), further indicating that Aβ1-42 induces removal of NCAM2 off the neuronal cell surface. In contrast, levels of the soluble proteolytic products of CHL1, another synaptic cell adhesion molecule of the immunoglobulin superfamily26, 27, were not changed in the culture medium from Aβ1-42-treated hippocampal neurons (Fig. 6b). Incubation with Aβ1-42 did not increase levels of soluble NCAM2 in the culture medium from cortical neurons (Fig. 6b), suggesting that cortical neurons are more resistant to Aβ1-42-dependent NCAM2 proteolysis.
Aβ binds to and removes NCAM2 from synapses in APP23 mice
Disruption of NCAM2 adhesion promotes synapse disassembly
Figure 9: Disruption of NCAM2 functions at the neuronal cell surface promotes glutamatergic synapse disassembly.

http://www.nature.com/ncomms/2015/151127/ncomms9836/images_article/ncomms9836-f9.jpg
(a–e) Cultured hippocampal neurons were either mock-treated or incubated with the recombinant soluble extracellular domains of NCAM2 (NCAM2-ED), antibodies against the extracellular domain of NCAM2 (NCAM2mAb), or Aβ1-42 oligomers. In a,b, neurons were labelled with antibodies against the extracellular domain of GluR1 before permeabilization of membranes with detergent, and co-labelled with antibodies against synaptophysin after permeabilization of membranes with detergent. Representative images of dendrites are shown (a). Note co-localization of cell surface GluR1 accumulations with synaptophysin clusters in mock-treated neurons, and increased levels of non-synaptic cell surface GluR1 accumulations in neurons treated with NCAM2-ED, NCAM2mAb or Aβ1-42. Graphs (b) show the percentage of synaptic and non-synaptic GluR1 clusters relative to total number of GluR1 clusters along dendrites and numbers of synaptophysin accumulations per dendrite length (mean+s.e.m.). *P<0.0001 (analysis of variance with Dunnett’s multiple comparison test, n>80 dendrites from 20 neurons were analysed in each group). In c, neurons were labelled with antibodies against the extracellular domain of NR1 before permeabilization of membranes with detergent, and co-labelled with antibodies against synaptophysin after permeabilization of membranes with detergent. Graphs show the percentage of synaptic and non-synaptic NR1 clusters relative to total number of NR1 clusters along dendrites (mean+s.e.m.). *P<0.0001 (analysis of variance with Dunnett’s multiple comparison test, n>85 dendrites from 20 neurons were analysed). In d,e, neurons were co-labelled with fluorescent phalloidin and synaptophysin antibodies. Representative images of dendrites are shown in d. Note higher labelling intensity and co-localization with synaptophysin of the phalloidin-labelled polymerized actin accumulations in control neurons versus neurons treated with Aβ1-42, NCAM2-ED or NCAM2mAb. Note increased numbers of filopodia and lamellipodia in neurons treated with Aβ1-42, NCAM2-ED or NCAM2 mAb. Graphs (e) show ratio of the dendrite area-to-length and phalloidin labelling intensity of dendrites of neurons. Mean values+s.e.m. are shown. *P<0.0001 (analysis of variance with Dunnett’s multiple comparison test, n=50 dendrites from 20 neurons were analysed in each group). Scale bar, 10 μm (in a,d).
Cleavage-resistant NCAM2 reduces Aβ-dependent synapse loss
Figure 10: Aβ1-42 reduces the number of GluR1-containing synapses in the NCAM2-dependent manner.
http://www.nature.com/ncomms/2015/151127/ncomms9836/images_article/ncomms9836-f10.jpg
(a) Representative images of dendrites of cultured hippocampal neurons transfected either with control negative miRNA (negative miR) or NCAM2miR and either mock-treated or incubated with Aβ1-42. Transfected neurons were identified by fluorescence of GFP, which is co-expressed together with miRNA. Neurons were co-labelled with antibodies against cell surface GluR1 and synaptophysin. Note that the number of synaptic GluR1 clusters is reduced and the number of non-synaptic GluR1 clusters is increased in neurons transfected with NCAM2miR. Scale bar, 10 μm. (b,c) Graphs show mean+s.e.m. percentage of synaptic and non-synaptic GluR1 clusters relative to the total number of GluR1 clusters along dendrites (b) and numbers of synaptophysin accumulations per dendrite length normalized to the mean number in mock-treated neurons (c) for neurons described in (a). (d–f) Graphs show mean+s.e.m. percentage of synaptic and non-synaptic GluR1 clusters relative to the total number of GluR1 clusters along dendrites (d), number of synaptophysin accumulations per dendrite length normalized to the mean number in mock-treated neurons (e), and area/length ratio (f) in cultured hippocampal neurons transfected either with GFP alone or co-transfected with GFP and non-mutated NCAM2 (NCAM2WT) or NCAM2D693A mutant and either mock-treated or incubated with Aβ1-42. (g,h) Graphs show mean+s.e.m. percentage of non-synaptic GluR1 clusters relative to the total number of GluR1 clusters along dendrites (g) and area/length ratio (h) in cultured hippocampal neurons co-transfected with NCAM2 miR and either GFP, non-mutated NCAM2 (WT) or NCAM2D693A mutant (D693A) and either mock-treated or incubated with Aβ1-42. In b–h, *P<0.01 (compared as indicated), ˆP<0.01 (compared with mock-treated neurons transfected with negative miR (b), GFP (d–f) or co-transfected with NCAM2miR and GFP (g–h)), analysis of variance with Tukey’s multiple comparison test, n>50 dendrites from 20 neurons were analysed in each group.
Taken together, our results indicate that Aβ affects the numbers of GluR1-containing glutamatergic synapses in a NCAM2-dependent manner.
Alzheimer’s disease is characterized by loss of synapses, which is the strongest correlate of cognitive decline11, 29, 30, 31, 32 and possibly one of the earliest events in AD pathogenesis30, 33. Synapses are long lasting contacts between neurons, which are stabilized by a number of cell adhesion molecules that concentrate in pre- and postsynaptic membranes2, 5. Cell adhesion molecules play an essential role in maintaining synapse functionality and stability. Although cell adhesion molecules of many families are required for the synapse integrity8, 10, elimination of even one type of synaptic cell adhesion molecule is often sufficient to induce abnormalities in synapse ultrastructure and protein composition6, 7. In the present study, we show that levels of the synaptic cell adhesion molecule NCAM2 are markedly reduced in hippocampal synapses in AD brains and Aβ-forming APP23 mice. Our observations that disruption of NCAM2 interactions at the cell surface, knockdown of NCAM2 expression and Aβ exposure result in reduced numbers of glutamatergic synapses in hippocampal neurons suggest that abnormalities in NCAM2-mediated synaptic adhesion contribute to synapse loss in AD.
Although the mechanisms of synapse disassembly in AD remain poorly understood, previous studies indicated that synapse loss can be linked to Aβ-induced toxicity12, 34, 35. Our observations showing that synaptic levels of NCAM2 are similarly reduced in APP23 mice and in cultured hippocampal neurons from wild-type mice exposed to Aβ argue in favour of Aβ-dependent mechanisms in the disruption of NCAM2-mediated synaptic adhesion. We however do not exclude that other factors, such as disrupted trafficking of NCAM2 to synapses, may also contribute to the reduction of NCAM2 levels at synapses. Strikingly, the effects of Aβ on synaptic targeting of NCAM2 were particularly strong in hippocampal but not cortical or cerebellar neurons. The enhanced susceptibility of synaptic NCAM2 to Aβ-dependent proteolysis may therefore contribute to selective vulnerability of the hippocampus to AD.
Our observations that NCAM2 directly interacts with synthetic Aβ1-42, that Aβ1-42 forms a molecular complex with NCAM2 at the neuronal cell surface and that complexes of NCAM2 and oligomers of Aβ can be isolated from APP23 mouse brains, indicate that NCAM2 may function as a previously unrecognized receptor for Aβ at the neuronal cell surface. Previous studies have shown that Aβ can also bind to other cell adhesion molecules at the neuronal cell surface, among which are the prion protein36 and L137. In addition, a number of cell adhesion molecules have been shown to interact with APP, including the neural cell adhesion molecule 1 (NCAM1)38 and TAG1 (ref. 39). It remains to be investigated whether the NCAM2/Aβ complex comprises other adhesion molecules and cell surface proteins. Interestingly, NCAM1, a homologue of NCAM2, binds to prion protein40 and L1 (ref. 41). However, in spite of homology to NCAM2, NCAM1 binds to a region of APP which is different to the Aβ-containing region38.
…..
Taken together, we show that Aβ induces synaptic loss and proteolysis of NCAM2 in cell culture and APP transgenic mouse models, providing a mechanistic explanation for synaptic NCAM2 changes in AD brains. The detrimental effects of proteolyically cleaved extracellular NCAM2 on synapses may augment the Aβ toxicity in the pathogenesis of AD. The exact molecular mechanisms underlying Aβ-induced NCAM2 changes, and to which degree it contributes to onset and progression of disease remains to be established. Nevertheless, our data reveal a new role of NCAM2 in AD that warrants further investigation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}