Regulation of mesenchymal cell generation
Curator: Larry H. Bernstein, MD, FCAP
from Butyrov
LPBI
Controlling Mesenchymal Stem Cell Activity With Microparticles Loaded With Small Molecules
M. Butyrov Beyond the Dish
Mesenchymal stem cells are the subject of many clinical trials and show a potent ability to down-regulate unwanted immune responses and quell inflammation. A genuine challenge with mesenchymal stem cells (MSCs) is controlling the genes they express and the proteins they secrete.
A new publication details the strategy of one enterprising laboratory to control MSC function. Work by Jeffery Karp from the Harvard Stem Cell Institute and Maneesha Inamdar from the Institute for Stem Cell Biology and Regenerative Medicine in Bangalore, India and their colleagues had use microparticles that are loaded with small molecules and are readily taken up by cultures MSCs.
In this paper, which appeared in Stem Cell Reports (DOI:http://dx.doi.org/10.1016/j.stemcr.2016.05.003), human MSCs were stimulated with a small signaling protein called Tumor Necrosis Factor-alpha (TNF-alpha). TNF-alpha makes MSCs “angry” and they pour out pro-inflammatory molecules upon stimulation with TNF-alpha. However, to these TNF-alpha-stimulated, MSC, Karp and others added tiny microparticles loaded with a small molecule called TPCA-1. TPCA-1 inhibits the NF-κB signaling pathway, which is one of the major signal transduction pathways involved in inflammation.
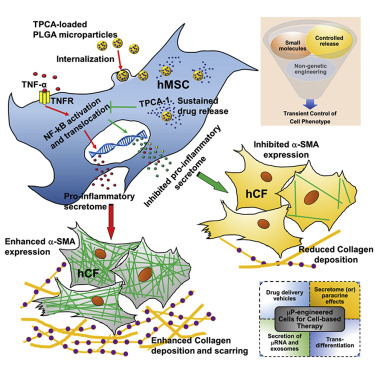
Delivery of these TPCA-1-containing microparticles thinned-out the production of pro-inflammatory molecules by these TNF-alpha-treated MSCs for at least 6 days. When the culture medium from TPCA-1-loaded MSCs was given to different cell types, the molecules secreted by these cells reduced the recruitment of white blood cells called monocytes. This is indicative of the anti-inflammatory nature of TPCA-1-treated MSCs. The culture medium from these cells also prevented the differentiation of human cardiac fibroblasts into collagen-making cells called “myofibroblasts.” Myofibroblasts lay down the collagen that produces the heart scar after a heart attack. This is a further indication of the anti-inflammatory nature of the molecules made by these TPCA-1-treated MSCs.
These results are important because it shows that MSC activities can be manipulated without gene therapy. It is possible that such non-gene therapy-based approached can be used to fine-tune MSC activity and the types of molecules secreted by implanted MSCs. Furthermore, given the effect of these cells on monocytes and cardiac fibroblasts, perhaps microparticle-treated MSCs can prevent the adverse remodeling that occurs in the heart after a heart attack.
Controlled Inhibition of the Mesenchymal Stromal Cell Pro-inflammatory Secretome via Microparticle Engineering
Mesenchymal stromal cells (MSCs) are promising therapeutic candidates given their potent immunomodulatory and anti-inflammatory secretome. However, controlling the MSC secretome post-transplantation is considered a major challenge that hinders their clinical efficacy. To address this, we used a microparticle-based engineering approach to non-genetically modulate pro-inflammatory pathways in human MSCs (hMSCs) under simulated inflammatory conditions. Here we show that microparticles loaded with TPCA-1, a smallmolecule NF-kB inhibitor, when delivered to hMSCs can attenuate secretion of pro-inflammatory factors for at least 6 days in vitro. Conditioned medium (CM) derived from TPCA-1-loaded hMSCs also showed reduced ability to attract human monocytes and prevented differentiation of human cardiac fibroblasts to myofibroblasts, compared with CM from untreated or TPCA-1-preconditioned hMSCs. Thus, we provide a broadly applicable bioengineering solution to facilitate intracellular sustained release of agents that modulate signaling. We propose that this approach could be harnessed to improve control over MSC secretome post-transplantation, especially to prevent adverse remodeling post-myocardial infarction.
Mesenchymal stromal cells (MSCs; also known as bone marrow stromal cells and earlier known as mesenchymal stem cells) are being explored as therapeutics in over 550 clinical trials registered with the US Food and Drug Administration (www.FDA.gov) for the treatment of a wide range of diseases (Ankrum et al., 2014c). Their immuneevasive properties (Ankrum et al., 2014c) and safe transplant record, allowing allogeneic administration without an immunosuppressive regimen, positions MSCs as an appealing candidate for a potential off-the-shelf product. One of the primary mechanisms exploited in MSC therapeutics is a secretome-based paracrine effect as evidenced in many pre-clinical studies (Ranganath et al., 2012). However, controlling the MSC secretome post-transplantation is considered a major challenge that hinders their clinical efficacy. For instance, upon transplantation, MSCs are subjected to a complex inflammatory milieu (soluble mediators and immune cells) in most injury settings. MSCs not only secrete anti-inflammatory factors, but also produce pro-inflammatory factors that may compromise their therapeutic efficacy. Table S1 lists a few in vitro and in vivo conditions that demonstrate the complex microenvironment under which MSCs switch between anti-inflammatory and pro-inflammatory phenotypes.
Levels of pro- or anti-inflammatory cytokines are not always predictive of the response, possibly due to the dynamic cytokine combinations (and concentrations) present in the cell microenvironment (Table S1). For example, relatively low inflammatory stimulus (<20 ng/ml tumor necrosis factor alpha [TNF-a] alone or along with interferon-g [IFN-g]) can polarize MSCs toward pro-inflammatory effects (Bernardo and Fibbe, 2013) resulting in increased inflammation characterized by T cell proliferation and transplant rejection. Conversely, exposure to high levels of the inflammatory cytokine TNF-a has been shown in certain studies to result in MSC-mediated anti-inflammatory effects via secretion of potent mediators such as TSG6, PGE2, STC-1, IL-1Ra, and sTNFR1 as demonstrated in multiple inflammation-associated disease models (Prockop and Oh, 2012; Ylostalo et al., 2012). These effects are mediated via molecular pathways such as NF-kB, PI3K, Akt, and JAK-STAT (Ranganath et al., 2012). However, it is not clear that low and high levels of TNF-a always exert the same effect on anti- versus pro-inflammatory MSC secretome. NF-kB is a central regulator of the anti-inflammatory secretome response in monolayer (Yagi et al., 2010), spheroid MSCs (Bartosh et al., 2013; Ylostalo et al., 2012) and TNFa-mediated (20 ng/ml for 120 min) apoptosis (Peng et al., 2011). Given that NF-kB can promote secretion of proinflammatory components in the MSC secretome (Lee et al., 2010), we hypothesized that NF-kB inhibition via small molecules in MSC subjected to a representative in- flammatory stimulus (10 ng/ml TNF-a) would inhibit their pro-inflammatory responses.
Adverse remodeling or cardiac fibrosis due to differentiation of cardiac fibroblasts (CF) into cardiac myofibroblasts (CMF) with pro-inflammatory phenotype and collagen deposition is the leading cause for heart failure. The secretome from exogenous MSCs has anti-fibrotic and angiogenic effects that can reduce scar formation (Preda et al., 2014) and improve ejection fraction when administered early or prior to adverse remodeling (Preda et al., 2014; Tang et al., 2010; Williams et al., 2013). Unfortunately in many cases, due to poor prognosis, MSCs may not be administered in time to prevent adverse remodeling to inhibit CF differentiation to CMF (Virag and Murry, 2003) or to prevent myocardial expression of TNF-a (Bozkurt et al., 1998; Mann, 2001). Also, when administered following an adverse remodeling event including CMF differentiation, MSCs may assume a pro-inflammatory phenotype and secretome (Naftali-Shani et al., 2013) under TNF-a (typically 5 pg/mg of total protein in myocardial infarction (MI) rat myocardium which is not significantly higher than 1–3 pg/mg protein in control rat myocardium) (Moro et al., 2007) resulting in impaired heart function. Hence, if the pro-inflammatory response of hMSCs can be suppressed it may maximize efficacy.
While the MSC phenotype can be controlled under regulated conditions in vitro, the in vivo response of MSCs post-transplantation is poorly controlled as it is dictated by highly dynamic and complex host microenvironments (Discher et al., 2009; Rodrigues et al., 2010). Factors including MSC tissue source (Melief et al., 2013; NaftaliShani et al., 2013), donor and batch-to-batch variability with respect to cytokine secretion and response to inflammatory stimuli (Zhukareva et al., 2010), gender (Crisostomo et al., 2007), and age (Liang et al., 2013) also affect the response of MSCs. Thus, it is important to develop approaches to control the MSC secretome post-transplantation regardless of their source or expansion conditions. We hypothesized that engineering MSCs to induce a specific secretome profile under a simulated host microenvironment may maximize their therapeutic utility. Previously, the MSC secretome has been regulated via genetic engineering (Gnecchi et al., 2006; Wang et al., 2009) or cytokine/small-molecule preconditioning approaches (Crisostomo et al., 2008; Mias et al., 2008). Genetically engineered human MSCs (hMSCs) pose challenging long-term regulatory hurdles given that the potential tumorigenicity has not been well characterized, and while preconditioning hMSCs with cytokines/small molecules may be safer, the phenotype-altering effects are transient. …
While TPCA-1 pretreatment of hMSCs before TNF-a stimulation showed a significant reduction in CMF numbers, this was greatly enhanced when TPCA-1 was available intracellularly via the microparticles. CM from TNF + TPCAmP-hMSC likely prevented differentiation of CF to CMF due to the continued intracellular inhibition of IKK-mediated NF-kB activation, thus preventing the release of an hMSC pro-inflammatory secretome. Surprisingly, the CMF number was reduced by over 2-fold, suggesting that TPCA-1 may activate hMSC pathways that revert the CMF phenotype. We cannot rule out the possibility that other contributors in the hMSC secretome that were not profiled might also be contributing, and their action is facilitated by intracellular TPCA-1. For instance, in an in vitro 3D model of cardiac fibrosis under hypoxic conditions, reversal of CMF to the CF phenotype was shown to be due to reduced MSC TGF-b levels (Galie and Stegemann, 2014). Our assay revealed a similar trend in terms of collagen production from CF. CF treated with CM from control-hMSCs, or TPCApre + TNF-hMSCs or mP-hMSCs secreted elevated levels of collagen into the media suggesting that only inhibited levels of pro-inflammatory mediators could not be implicated in the reduction in collagen secretion. The reduced number of a-SMA+ CMF in CM from TNF + TPCAmP-hMSCs possibly contributed to the lower secretion level of collagen in the media (Figure 5B). Upon dedifferentiation or lowered a-SMA expression, it is possible that CMFs lose collagen secretion ability. In regions of MI, CF switch to the myofibroblast phenotype due to stress from the infarct scar (Tomasek et al., 2002). High expression of a-SMA typical in such CMF (Teunissen et al., 2007) has been implicated for remodeling due to their high contractility (Santiago et al., 2010). In addition, the collagen secretion capacity of CMF is very high (Petrov et al., 2002). Overall, attenuation in the number of collagen-secreting a-SMA+ CMF could be beneficial in preventing pathological remodeling or irreversible scar formation and allowing cardiac regeneration.
Here we have demonstrated that the pro-inflammatory hMSC secretome could be inhibited using a microparticle engineering approach delivering an intracellular NF-kB inhibitor, TPCA-1. It is however important to note that the MSC secretome composition may change depending on the level of TNF-a encountered in vivo following transplantation. Nevertheless, a similar approach could be beneficial in inflammatory disease settings such as chronic inflammation and macrophage-mediated atherosclerosis. The approach of microparticle engineering of an exogenous cell population by modulating a central regulatory pathway, may find application in other cell types and pathways and could provide an attractive strategy for harnessing any cell secretome for therapy. This approach could also be potentially employed to modulate the composition of extracellular vesicles (exosomes) for therapy.
Stem Cell Reports j Vol. 6 j 926–939 j June 14, 2016
Read Full Post »


{kind=link}