The Final Considerations of the Role of Platelets and Platelet Endothelial Reactions in Atherosclerosis and Novel Treatments
Author and Curator: Larry H Bernstein, MD, FCAP
Introduction
We have seen the emergence of novel antiplatelet medications for the treatment of patients who undergo cardiovascular interventions. This came as a dramatic escalation in research-based knowledge about platelet function, about endothelial-platelet interactions, about platelet induced protein synthesis, and about cell receptors as targets of therapy. A significant part of the story has been presented in several articles. There is yet more that has been done with potential for further progress in the treatment of cardiovascular disease.
An abbreviated list of articles published in Pharmaceutical Intelligence so far is:
Platelets in Translational Research – 1
http://pharmaceuticalintelligence.com/10-6-2013/larryhbern/Platelets_in_Translational_Research-1
Platelets in Translational Research – 2
http://phramaceuticalintelligence.com/2013-10-7/larryhbern/Platelets-in-Translational-Research-2/
αllbβ3 Antagonists As An Example of Translational Medicine Therapeutics
http://phrmaceuticalintelligence.com/2013-10-12/larryhbern_BS-Coller/αllbβ3_Antagonists_As_An_Example_of_Translational_Medicine_Therapeutics
Do Novel Anticoagulants Affect the PT/INR? The Cases of XARELTO (rivaroxaban) and PRADAXA (dabigatran)
Vivek Lal, MBBS, MD, FCIR, Justin D Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN http://pharmaceuticalintelligence.com/2013/09/23/do-novel-anticoagulants-affect-the-ptinr-the-cases-of-xarelto-rivaroxaban-and-pradaxa-dabigatran/
Intravenous drug for the treatment of Acute Heart Failure (AHF) by Trevena, Inc. (Trevena) – Leader in the Discovery of G-protein coupled receptor (GPCR) biased ligands
Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/10/10/intravenous-drug-for-the-treatment-of-acute-heart-failure-ahf-by-trevena-inc-trevena-leader-in-the-discovery-of-g-protein-coupled-receptor-gpcr-biased-ligands/
Landscape of Cardiac Biomarkers for Improved Clinical Utilization
http://pharmaceuticalintelligence.com/2013/09/22/landscape-of-cardiac-biomarkers-for-improved-clinical-utilization/
Nitric Oxide, Platelets, Endothelium and Hemostasis
http://pharmaceuticalintelligence.com/2012/11/08/nitric-oxide-platelets-endothelium-and-hemostasis/
Nitric Oxide Function in Coagulation
http://pharmaceuticalintelligence.com/2012/11/26/nitric-oxide-function-in-coagulation/
Platelet-endothelial interactions in atherosclerosis
Bruce S. Sachais MD, PhD
http://link.springer.com/article/10.1007/s11883-001-0080-1
Current Atherosclerosis Reports 2001; 3(5): 412-416
The pathogenesis of atherosclerosis, the leading cause of morbidity and mortality in the United States, is multifactorial. Many factors that have been shown to influence the development of atherosclerosis also affect the function of the endothelium through soluble or cell-cell interactions. Among these, interactions between platelets and endothelial cells have only recently begun to receive systematic study. This article reviews recent evidence showing how the interaction between platelets and endothelial cells may play a important role in the pathogenesis of atherosclerosis, suggesting an underappreciated potential locus for pharmacologic intervention.
http://link.springer.com/article/10.1007%2Fs11883-001-0080-1/lookinside/000.png Date 2001-09-01
Online ISSN 1534-6242 http://dx.doi.org/10.1007/s11883-001-0080-1
{kind=link}
Rosuvastatin reverses plaque build-up in atherosclerosis
http://link.springer.com/article/10.2165/00128413-200615290-00019
Inpharma Weekly Mar 2006; 1529(1): 10
http://dx.doi.org/10.2165%2F00128413-200615290-00019
Semin Thromb Hemost. 2007 Mar;33(2):128-35.
Platelets and endothelial cells.
http://www.ncbi.nlm.nih.gov/pubmed/17340460
Medizinische Klinik III, Eberhard Karls-Universität Tübingen, Tübingen, Germany
Atherogenesis, the development of atherosclerotic lesions, is initiated by a mechanical or functional injury of the endothelium. The function of the endothelium is influenced by multiple factors as a consequence of cell-cell interactions. Cell-cell communication between endothelial cells with platelets has only recently begun to receive systematic study. In recent years it has been established that platelet-endothelial interactions are involved at all stages of atherosclerotic disease. This article reviews the interactions between endothelial cells and platelets in the context of their role to initiate and accelerate atherothrombosis, as well as in acutethrombotic occlusion (e.g., at the site of atherosclerotic plaque rupture or subsequent to coronary angioplasty). From a mechanistic standpoint, platelets and endothelial cells communicate on multiple levels. Cross-talk may occur over a distance (paracrine signaling), via transient interactions (so-called give-and-go mechanism), or through receptor-mediated cell-cell adhesion. Platelets may release or transfer substances that influence endothelial cell function, and vice versa. Among many others, adhesion molecules, such as P-selectin (CD62P), are of special interest because of their role in modulating interactions between blood cells and the endothelium, and also because of the possible use of the soluble form as a plasma predictor of adverse cardiovascular events. In addition to dietary, cholesterol and lipid lowering, and other pharmaceutical approaches, antiplatelet therapy plays an important part in the treatment of atherosclerosis and its multifactorial clinical manifestations. Understanding the specific interactions between platelets and the endothelium may lead to the development of novel therapeutic strategies. PMID: 17340460
Platelets and atherogenesis: Platelet anti-aggregation activity and endothelial protection from tomatoes (Solanum lycopersicum L.).
Palomo I, Fuentes E, Padró T, Badimon L.
Depart Clin Biochem and Immunohem, Faculty of Health Sciences, University of Talca.
Exp Ther Med. 2012 Apr;3(4):577-584. Epub 2012 Feb 9. PMID: 22969932 PMCID: PMC3438755
http://www.ncbi.nlm.nih.gov/pubmed/22969932
In recent years, it has been shown that platelets are not only involved in the arterial thrombotic process, but also that they play an active role in the inflammatory process of atherogenesis from the beginning. The interaction between platelets and endothelial cells occurs in two manners:
- activated platelets unite with intact endothelial cells, or
- platelets in resting adhere to activated endothelium.
In this context, inhibition of the platelet function (adhesion/aggregation) could contribute to the prevention of atherothrombosis, the leading cause of cardiovascular morbidity. This can be achieved with antiplatelet agents. However, at the public health level, the level of primary prevention, a healthy diet has also been shown to exert beneficial effects. Among those elements of a healthy diet, the consumption of tomatoes (Solanum lycopersicum L.) stands out for its effect on platelet anti-aggregation activity and endothelial protection, which may be beneficial for cardiovascular health. This article briefly discusses the involvement of platelets in atherogenesis and the possible mechanisms of action provided by tomatoes for platelet anti-aggregation activity and endothelial protection.
According to the World Health Organization (WHO), cardiovascular disease (CVD) (i.e., acute myocardial infarction, cerebrovascular disease and peripheral arterial thrombosis) is the cause of approximately 30% of deaths worldwide, with a relative increase over time due to the aging of the population. The development and progression of CVD lies in the interactive processes of atherosclerotic lesions and thrombus formation, an interaction established primarily by platelet-endothelial binding
The activation of vascular endothelium occurs early in the development of atherosclerosis, where the inflammatory component, present in all phases of atherosclerosis, is a vascular response to guard against cardiovascular risk factors (i.e., hypertension, diabetes, smoking and obesity). This inflammatory process generates a microenvironment characterized by oxidative stress and cell damage. The process triggers a loss of endothelial function through a decrease in the bioavailability of nitric oxide (NO) and the physiological mechanisms of cardiovascular protection that are derived from it.
The role of platelets in arterial thrombosis is well known. When there is a damaged atheromatous plaque, platelets adhere, secrete their contents, and then attach to it. This activation causes a redistribution of anionic phospholipids, creating a negatively charged surface which, in addition to the synthesis and expression of tissue factor, favors the consecutive formation of protein complexes in coagulation and fibrin and the consolidation of the thrombus.
Epidemiological studies have demonstrated the cardiovascular protective role of a healthy diet. In this context, the beneficial effects of fruits and vegetables (F&V) may be related to the bioactive compounds found therein, which explains the increasing amount of attention in research on phytochemicals in the prevention of CVD. In addition to their nutritional value, tomatoes (Solanum lycopersicum L.), fresh or processed, have been found to provide a cardioprotective effect at both the endothelial and platelet levels.
This article discusses current knowledge of platelet-endothelial interaction during the development of atherosclerosis, and platelet anti-aggregation activity and the endothelial protective effects from tomatoes.
Endothelium in the normal state plays a fundamental role in regulating the hemostatic balance through various mechanisms of antiplatelets, anticoagulants and fibrinolysis, which are regulated by the secretion of NO and prostacyclin.
In vitro studies have demonstrated platelet adhesion to ECs in the normal state. Platelet adhesion occurs because it is activated in circulation. By contrast, in other in vivo studies, the binding occurs under shear conditions. Once activated, platelets may adhere to ECs and promote local vascular inflammation through inflammatory mediators such as the secretion of chemokines, which disrupt normal functioning of the endothelium . For example, platelets store and express CD40L (inflammatory modulator) on their surface, releasing the protein into the environment once they are activated. CD40L also induces the expression and release of metalloproteinases, which degrade extracellular matrix proteins that are exposed to the circulation when damage occurs at the endothelial level. In addition to the release of sCD40, IL-1β, which promotes increased IL-6 and IL-8 and the expression of cell adhesion molecules such as E-selectin, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in the EC, is also released. This leads to the recruitment of leukocytes to the site from which the injury (i.e., damaged endothelium) originated. The leukocyte-platelet interaction causes a wide range of responses in the innate and adaptive immune systems, giving the platelet a new function as regulator of the immune system, which contributes to the pathogenesis of an inflammatory response.
Moreover, animal models have shown a high level of P-selectin expression and endothelial growth factor (VEGF) in atherosclerotic plaques with involvement in the progression of CVD. Platelet factor 4 (PF-4), CCL5 (RANTES) and platelet-derived growth factor (PDGF) molecules released from activated platelets cause chemotaxis of monocytes and other leukocytes on the EC, and promote the retention of low density lipoprotein (LDL) and oxidized LDL in the subendothelium. In patients with CVD, the presence of oxidized LDL promotes the release of neutrophil-peptide 78 (ENA-78), with an increase in chemoattractant capacity. The deposit of RANTES on the endothelium is promoted by platelet P-selectin. PDGF also stimulates smooth muscle cell proliferation, causing hyperplasia of the intima layer of the arterial wall, thus acting as an amplifier of the inflammatory response.
It has been demonstrated that ADAM-15 expressed in activated ECs interacts with the GPIIb/IIIa complex and induces platelet activation. ADAM-15 also operates as a metalloproteinase, degrading endothelial matrix proteins. The two functions of this molecule could facilitate the instability and rupture of the fibrous cap of atherosclerotic lesions, causing the acceleration of the formation of a platelet thrombus.
Tomatoes: platelet anti-aggregation activity and endothelial protection
The report ‘Diet, Nutrition and the Prevention of Chronic Diseases’, published by the WHO in 2003, outlined the scientific evidence that has been associated with a decreased risk of CVD. According to the report, individuals who consume at least 400 g of F&V daily were found to have a reduced risk of CVD. By consuming 5 servings of F&V daily there is a 17% reduction in the risk of CVD. Furthermore, the effects of consuming F&V have been replicated worldwide and are independent of ethnicity or geographical location.
Presently, in addition to their recognized high value in vitamins, minerals and dietary fiber, consuming F&V is associated with phytochemical content, with specific actions on target functions. These effects, in terms of primary prevention, could modify cardiovascular risk without any of the side effects normally associated with the majority of antiplatelet drugs.
The antioxidant properties of F&V are well known. However, their antithrombotic effects on platelet-endothelial interaction are less known. Preliminary studies have demonstrated the platelet anti-aggregation activity of fruits (red grapes, strawberries, kiwis and pineapples) and vegetables (garlic, onions, green onions, melons and tomatoes). Among those beneficial elements mentioned above, the consumption of tomatoes, the fruit of a dicotyledonous plant belonging to the Solanaceae family, is emphasized. The possible mechanisms of action of bioactive compounds from tomatoes that have platelet anti-aggregation activity and endothelial protective effects are subsequently discussed.
It has been observed that the tomato has platelet anti-aggregation activity in vitro and in vivo by inhibiting platelet aggregation induced by ADP and collagen. This finding has also been confirmed by our research group (71). The various platelet anti-aggregation activity levels observed in different varieties of tomatoes can be explained by the existence of one or more bioactive compounds or different concentrations of the same compound. The platelet anti-aggregation activity of aqueous and methanol extracts of tomatoes (‘cluster’ type) in vitro were similar. Both types of extract showed inhibition of platelet aggregation (30–40%) at 1 mg/ ml induced by ADP. When collagen was used as agonist, inhibition was lower, whereas the use of arachidonic acid and peptide receptor activator of thrombin showed no inhibitory effect.
The experimental results obtained by Fuentes et al indicate that aqueous and methanol extracts resuspended in 0.9% saline exhibit a pH of 4.5; when resuspended in more acidic (pH 2.0) and basic (pH 10.0) suspensions, they maintained their inhibitory activity of maximum platelet aggregation. As we know that carotenoids are unstable at pH extremes, this finding may exclude the possibility that these antioxidant compounds exhibit platelet anti-aggregation activity. In addition, the platelet anti-aggregation activity is inversely related to tomato ripening and the increase in the concentration of lycopene. The active compounds with platelet anti-aggregation activity present in the two extracts were not affected by heat treatment.
Lycopene is considered to be a chemopreventive agent, as it maintains the integrity of the vascular barrier, inhibits the expression of cell adhesion molecules and leukocytes, and inhibits EC migration by blocking the expression of NF-κB, CD14 and TLR4, and TNF-α production. In addition to decreased levels of malondialdehyde, programmed cell death prevents apoptosis by attenuating the expression of p53 and caspase-3 in ECs treated with H2O2.
Newly discovered mechanisms include: the reduction of endothelial injury, control of lipid metabolism during the synthesis of cholesterol and oxysterol toxic activity and reduction of the inflammatory response through changes in cytokine production. While it is know that the consumption of tomato products is associated with a significant increase in plasma levels of lycopene, this has no substantial effect on endothelial function. Studies have also found that there are other potent bioactive compounds in tomatoes, such as flavonoids, whose nanomolar concentrations protect the cofactor tetrahydrobiopterin from peroxynitrite radicals and maintain the action of endothelial nitric oxide synthase (eNOS). The inhibition of arginase enzyme and of NADPH oxidase combined with O2, which causes a positive NO balance in the EC and prevents apoptosis through p53, has also been shown to inhibit the synthesis of endothelian-1 (ET-1). Its immediate anti-apoptotic function is to block the JNK and p38 MAPK signaling pathways, and its resistance to LDL oxidation through ROS takes place through action on the JAK2/ STAT3 pathways.
To further understand the endothelial protective effects of bioactive compounds, such as those mentioned above, or those of other bioactive compounds that may be present in the tomato, future studies should focus on the three most important properties of the endothelium:
- markers of vascular tone control, i.e., concentrations of asymmetric dimethylarginine (ADMA), NO, eNOS and ET-1;
- ii) markers of the regulation of hemostasis, i.e., concentrations of prostacyclin, plasminogen activator inhibitor-1 (PAI-1) and tissue plasminogen activator (tPA); and
- iii) markers of the immune system, i.e., the presence of sICAM-1, sE-selectin, sVCAM-1, IL-6, TNF-α and CRP high-sensitivity (hsCRP).
4.Conclusion
The initiation and development of CVD is marked by platelet-endothelial interaction. This interaction promotes the expression of adhesion molecules on the endothelium and the recruitment of inflammatory cells, and stimulates the activation of circulating platelets. In the prevention of CVD, the consumption of F&V is crucial. At the level of primary prevention, tomato consumption promotes cardiovascular health through its role in platelet anti-aggregation activity and its endothelium-protective effects. Platelet anti-aggregation activity is regulated by one or more bioactive compounds that act on ADP and collagen receptors. Further research is required in the identification of mechanisms of action of bioactive compounds. In the endothelium, carotenoids and polyphenols act mainly on eNOS and NAPDH-oxidase in order to control the levels of NO and to ensure a reduction in the inflammatory response.
These results allow us to identify the tomato as a functional food, with one or more bioactive compounds with acid-base and thermal stability to exert its cardioprotective activity. This characteristic will benefit future efforts to protect the molecular structure and corresponding platelet anti-aggregation activity of tomato extracts during processing, storage, transport, management and molecular action. This will prove useful in the search for alternative antithrombotic therapy, a field in which most of the drugs used have a high instability in the environment of action.
References
Palomo I, Toro C, Alarcón M. The role of platelets in the pathophysiology of atherosclerosis (Review) Mol Med Rep. 2008;1:179–184. [PubMed]
Palomo I, Alarcón M, Moore-Carrasco R, Argilés J. Hemostasis alterations in metabolic syndrome (Review) Int J Mol Med. 2006;18:969–974. [PubMed]
Torres-Urrutia C, Guzmán L, Schmeda-Hirschmann G, et al. Antiplatelet, anticoagulant, and fibrinolytic activity in vitro of extracts from selected fruits and vegetables. Blood Coagul Fibrinolysis. 2011;22:197–205. [PubMed]
Dutta-Roy A, Crosbie L, Gordon M. Effects of tomato extract on human platelet aggregation in vitro. Platelets. 2001;12:218–227. [PubMed]
Yamamoto J, Taka T, Yamada K, et al. Tomatoes have natural anti-thrombotic effects. Br J Nutr. 2003;90:1031–1038. [PubMed]
Fuentes E, Astudillo L, Gutiérrez M, et al. Fractions of aqueous and methanolic extracts from tomato (Solanum lycopersicum L.) present platelet antiaggregant activity. Blood Coagul Fibrinolysis. 2012;23:109–117. [PubMed]
O’Kennedy N, Crosbie L, Whelan S, et al. Effects of tomato extract on platelet function: a double-blinded crossover study in healthy humans. Am J Clin Nutr. 2006;84:561–569. [PubMed]
Platelets: pleiotropic roles in atherogenesis and atherothrombosis.
Linden MD, Jackson DE.
School of Medical Sciences, RMIT University, Bundoora, VIC, Australia. matthew.linden@rmit.edu.au
Int J Biochem Cell Biol. Nov 2010;42(11):1762-6.
http://dx.doi.org/10.1016/j.biocel.2010.07.012. Epub 2010 Jul 29.
Growing evidence suggests that platelets are also important mediators of inflammation and play a central role in atherogenesis itself. Interactions between activated platelets, leukocytes and endothelial cells trigger autocrine and paracrine activation signals, resulting in leukocyte recruitment at and into the vascular wall. Direct physical interaction may contribute also, through platelet adhesion molecules assisting localization of monocytes to the site of atherogenesis and platelet granule release contributing to the chronic inflammatory milieu which leads to foam cell development and accelerated atherogenesis.
Recent studies have shown that antiplatelet therapy in animal models of accelerated atherogenesis can lead to decreased plaque size and improve plaque stability. This review examines the complexity of platelet function and the nature of interactions between activated platelets, leukocytes and endothelial cells. We focus on the growing body of evidence that platelets play a critical role in atherogenesis and contribute to progression of atherosclerosis. http://www.ncbi.nlm.nih.gov/pubmed/20673808 PMID: 20673808
Signal-Dependent Protein Synthesis by Activated Platelets: New Pathways to Altered
Phenotype and Function
Guy A. Zimmerman and Andrew S. Weyrich
Arterioscler Thromb Vasc Biol.2008;28:s17-s24 http://atvb.ahajournals.org/content/28/3/s17
http://dx.doi.org/10.1161/ATVBAHA.107.160218
Abstract—New biologic activities of platelets continue to be discovered, indicating that concepts of platelet function in hemostasis, thrombosis, and inflammation require reconsideration as new paradigms evolve. Studies done over 3 decades ago demonstrated that mature circulating platelets have protein synthetic capacity, but it was thought to be low level and inconsequential. In contrast, recent discoveries demonstrate that platelets synthesize protein products with important biologic activities in a rapid and sustained fashion in response to cellular activation.This process, termed signal-dependent translation, uses a constitutive transcriptome and specialized pathways, and can alter platelet phenotype and functions in a fashion that can have clinical relevance. Signal-dependent translation and consequent protein synthesis are examples of a diverse group of posttranscriptural mechanisms in activated platelets that are now being revealed. (Arterioscler Thromb Vasc Biol. 2008;28:s17-s24)
Key Words: platelets . translation . protein synthesis . transcriptome . proteome . Thrombosis
Introduction: New Paradigms at the Vascular Interface
Platelets are unique cellular effectors of hemostasis and thrombosis that act rapidly and in concert with coagulation proteins and with other vascular cells. The acute hemostatic functions of platelets are well known, have dominated the attention of the field for decades, and have been the foundation for discoveries that generated new molecular therapies.1,2 Rapid, immediate activation responses mediate platelet-dependent thrombosis in a variety of pathologic conditions,3 and pharmacological antiplatelet strategies are largely aimed at these events.4 Nevertheless, the focus on adhesion, aggregation, and secretion, and the view that platelets have a repertoire of activities primarily restricted to these acute processes, have also generated a central dogma that may inappropriately limit our view of their actions at the vascular interface and in other settings in health and disease.5 Clearly, our understanding of the molecular mechanisms by which platelets influence hemostasis, thrombosis, regulated and dysregulated inflammation, and neoplasia remains incomplete and continues to evolve. New paradigms are emerging as previously unrecognized pathways in platelets are identified, and unanticipated activities are characterized. In this regard, the current state of the field of platelet biology may be akin to that of endothelial cells several decades ago, when endothelium was thought by most investigators and physicians to have a limited range of responses; on the contrary, however, when this dogma was reexamined using new approaches that included primary culture of human endothe-lium,6 active participation of these cells in interactions with leukocytes7,8 and a variety of other previously unrecognized functions were discovered.9 If the comparison is accurate, new paradigms relevant to activities of platelets at the vascular interface are likely to be reported with some frequency.
Alternative and traditional views of selected features of platelet biology are listed in the Table. There is already considerable evidence for some of the alternative themes, such as inflammatory and immune activities of platelets,10–16 whereas others are less well explored and more speculative. The remainder of this review summarizes evidence for one such functional capability not generally recognized in platelets until recent discoveries revealed it: synthesis of new protein products in response to cellular activation (reviewed in references5,17). General dogma in the field has assumed that platelets have limited or no capacity for protein synthesis
Table. New Biology of Platelets: Traditional Paradigms May Be Insufficient to Understand Platelet Activities at the Vascular Interface
Traditional View Alternate View
Platelets are biologically simple because they are anucleate Platelets have specializations and biologic activities that are novel and complex. Some
and have a limited repertoire of responses. activities are yet to be discovered.
Platelets do not express new gene products. Platelets have diverse posttranscriptional mechanisms and use a transcriptome and
specialized pathways to modify their proteome, phenotype, and functions.
Platelets are short-acting cells in clots and damaged tissue. Platelets can be relatively long-lived and can mediate cell-cell interactions for many
hours after initial adhesion, aggregation, and secretion.
Platelets operate exclusively in the intravascular Platelets can influence critical events in the extravascular milieu in direct
compartment. and indirect fashions.
Curr Pharm Des. 2007;13(16):1685-91.Weyrich A, Cipollone F, Mezzetti A, Zimmerman G.
Eccles Institute of Human Genetics, Salt Lake City, Utah 84112, USA.
The functional interplay of platelets with leukocytes and endothelial cells is critical both in physiological conditions, such as host defense, wound repair and tissue healing, and in pathological conditions, such as thrombotic and inflammatory diseases. The understanding of the specific molecular links underlying these cellular interactions and the functional changes that they induce could suggest new targets for pharmacological intervention. In this review, we summarize some of the newly-recognized aspects of interaction between platelets and monocytes, neutrophils and endothelial cells, that are relevant to all of the phases of atherosclerosis progression, since the early steps of atherogenesis to plaque rupture and thrombosis, the main causes of acute coronary syndromes. http://www.ncbi.nlm.nih.gov/pubmed/17584098
PMID: 17584098 [PubMed – indexed for MEDLINE]
Weyrich A, Cipollone F, Mezzetti A, Zimmerman G.
Eccles Institute of Human Genetics, Salt Lake City, Utah 84112, USA.
Curr Pharm Des. 2007;13(16):1685-91.
The understanding of the specific molecular links underlying the functional interplay of platelets with leukocytes and endothelial cells is critical to understanding thrombotic and inflammatory diseases. These cellular interactions and the functional changes that they induce could suggest new targets for pharmacological intervention. In this review, we summarize some of the newly-recognized aspects of interaction between platelets and monocytes, neutrophils and endothelial cells, relevant to all of the phases of atherosclerosis progression, beginning with the early steps of atherogenesis to plaque rupture and thrombosis. http://www.ncbi.nlm.nih.gov/pubmed/17584098 PMID: 17584098
Increased levels of neutrophil-activating peptide-2 in acute coronary syndromes: possible role of platelet-mediated vascular inflammation.
Smith C, Damås JK, Otterdal K, Øie E, Sandberg WJ, Yndestad A, Waehre T, et al.
Research Institute for Internal Medicine, Rikshospitalet University Hospital, University of Oslo, Oslo, Norway
J Am Coll Cardiol. 2006 Oct 17;48(8):1591-9. Epub 2006 Sep 27.
Abstract
We sought to investigate the role of the CXC chemokine neutrophil-activating peptide-2 (NAP-2) in atherogenesis and plaque destabilization.
Chemokines are involved in atherogenesis, but the role of NAP-2 in atherosclerotic disorders is unclear. Based on its potential pro-atherogenic properties, we hypothesized a pathogenic role for NAP-2 in coronary artery disease.
We tested this hypothesis by differential experimental approaches including studies in patients with stable (n = 40) and unstable angina (n = 40) and healthy control subjects (n = 20).
The following results were discovered:
-
patients with stable, and particularly those with unstable, angina had markedly raised plasma levels of NAP-2 compared with control subjects, accompanied by increased expression of CXC receptor 2 in monocytes;
-
platelets, but also peripheral blood mononuclear cells (PBMCs), released large amounts of NAP-2 upon stimulation, with a particularly prominent PBMC response in unstable angina;
-
NAP-2 protein was detected in macrophages and smooth muscle cells of atherosclerotic plaques and in monocytes and platelets of coronary thrombi;
-
in vitro, recombinant and platelet-derived NAP-2 increased the expression of adhesion molecules and chemokines in endothelial cells; and
-
aspirin reduced plasma levels of NAP-2,
-
statin therapy increased NAP-2 with stimulating effects both on platelets and leukocytes.
Our findings suggest that NAP-2 has the potential to induce inflammatory responses within the atherosclerotic plaque. By its ability to promote leukocyte and endothelial cell activation, such a
NAP-2-driven inflammation could promote plaque rupture and acute coronary syndromes. http://www.ncbi.nlm.nih.gov/pubmed/17045893PMID: 17045893
Pluripotential mechanisms of cardioprotection with HMG-CoA reductase inhibitor therapy.
Rosenson RS.
Preventive Cardiology Center, Northwestern University Medical School, Chicago, IL
Abstract
Rapid onset of clinical benefit from treatment with hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors has been accompanied by a reduced risk of cardiovascular events, but weak correlations between plasma low density lipoprotein-cholesterol levels and coronary lumen change or cardiovascular events indicates that nonlipid mechanisms are involved in beneficial effects with HMG-CoA reductase inhibitors.
The rapid onset of clinical benefit with HMG-CoA reductase inhibitors suggest HMG-CoA reductase inhibitors
- facilitate repair of ruptured or ulcerated atherosclerotic plaque,
- facilitate plaque stabilization and/or reduce thrombus formation on ruptured plaques.
Treatment with HMG-CoA reductase inhibitors
- improved endothelial dysfunction in patients with hypercholesterolemia and
- the improvement was not correlated with reduction in total serum cholesterol levels.
Similarly, reduction in endothelial pre-proendothelin mRNA expression and endothelin synthesis and blood pressure lowering with HMG-CoA reductase inhibitors occurred independent of lipid-lowering.
Finally, HMG-CoA reductase inhibitors increased endothelial nitric oxide levels i.e.
- upregulated endothelial nitric oxide synthetase expression via post-transcriptional mechanisms and
- prevented its down-regulation by oxidized LDL-C.
HMG-CoA reductase inhibitors have been shown to modulate the immune response by inhibiting activation of immune-competent cells such as macrophages, and antigen presentation to macrophages by T cells. Treatment with HMG-CoA reductase inhibitors can reduce expression, production and circulating levels of chemokines (monocyte chemoattractant protein-1) and proinflammatory cytokines [tumor necrosis factoralpha, interleukin (IL)-6 and IL-1beta]. HMG-CoA reductase inhibitors reduced inflammation in human atheroma:
- significantly fewer macrophages and T cells,
- less oxidized LDL-C and higher collagen content.
In addition, treatment with HMG-CoA reductase inhibitor led to decreased cell death within the atheroma. Treatment with these agents also
- reduced expression of inducible cellular adhesion molecules,
- decreased secretion of metalloproteinases by macrophages,
- reduced vascular smooth muscle cell apoptosis.
Lastly, HMG-CoA reductase inhibitors appear to have important effects on the thrombogenesis:
- reduced expression of tissue factor production and activity;
- increased production of tissue factor package inhibitor;
- decreased platelet thrombus formation and improved fibrinolysis as a result of lowered plasminogen activator inhibitor-1 levels.
As the pluripotential cardioprotective mechanisms of HMG-CoA reductase inhibitors are further elucidated, it is envisaged that treatment with HMG-CoA reductase inhibitors will be initiated
earlier and more frequently in patients with hypercholesterolemia. http://www.ncbi.nlm.nih.gov/pubmed/14728000PMID: 14728000
Effects of HMG-CoA reductase inhibitors on coagulation and fibrinolysis processes.
Krysiak R, Okopień B, Herman Z.
Department of Clinical Pharmacology, Medical University of Silesia, Medyków 18, Katowice, Poland.
Drugs. 2003;63(17):1821-54.
Abstract
Large clinical trials have demonstrated that HMG-CoA reductase inhibitors, or statins, markedly reduce morbidity and mortality when used in the primary and secondary prevention of cardiovascular disease. It has been established that the benefits of statin therapy in cardiovascular disease can be explained not only by the lipid-lowering potential of statins but also by nonlipid-related mechanisms (so-called “pleiotropic effects”).
The coagulation and fibrinolytic systems are two separate but reciprocally linked enzyme cascades that regulate the formation and breakdown of fibrin. Numerous studies have demonstrated that disturbances of coagulation and fibrinolysis contribute to the development and progression of atherosclerosis, and that they affect the incidence of atherosclerosis-related clinical events. High plasma levels or activities of fibrinogen, factor VII, factor VIII, von Willebrand factor (vWF), soluble thrombomodulin, tissue plasminogen activator (tPA) and plasminogen activator inhibitor-1 (PAI-1) are thought to be associated with increased morbidity and mortality related to cardiovascular disease. Experimental studies and many clinical studies have recently shown that statins produce favourable effects on haemostatic parameters, including those that are risk factors for cardiovascular disease.
Statins diminish procoagulant activity, which is observed at different stages of the coagulation cascade, including tissue factor (TF) activity, conversion of prothrombin to thrombin and thrombin activity. In some studies, statins also reduced fibrinogen levels. By altering the levels and activities of tPA and PAI-1, statins seem to stimulate fibrinolysis. The data on the effects of combined treatment with statins and other drugs on haemostasis are rather limited.
They suggest that statins combined with fibric acid derivatives, omega-3 fatty acids and 17beta-estradiol are superior to statins alone. The only two clinical studies performed in patients with acute coronary syndromes showed a relatively weak effect of statins on haemostasis in those patients. Although various statins may produce different effects on individual variables, there are no convincing data showing that differences in their physicochemical and pharmacokinetic properties significantly alter their net effect on excessive procoagulant activity.
Apart from the lipid-lowering effect, statins suppress the synthesis of several important nonsterol isoprenoids derived from the mevalonate pathway, especially farnesyl and geranylgeranyl pyrophosphates, which via enhanced protein prenylation, are involved in the regulation of many cellular processes. It is presumed that the inhibitory effect of statins on the mevalonate pathway is involved in the regulation of some key steps of coagulation and fibrinolysis processes. In this way they probably regulate the synthesis of TF, tPA and PAI-1, and perhaps they also control the generation and activity of thrombin. The beneficial effects of statins on coagulation and fibrinolysis may be responsible for their ability to decrease the number of cardiovascular events. The lipid-independent effects of statins on haemostasis may contribute to the marked decrease in the incidence rates of mortality, hospitalisation and revascularisation in patients treated with these drugs. http://www.ncbi.nlm.nih.gov/pubmed/12921488PMID: 12921488
Effects of short-term atorvastatin treatment on global fibrinolytic capacity, and sL-selectin and sFas levels in hyperlipidemic patients with coronary artery disease.
Atalar E, Ozmen F, Haznedaroglu I, Açil T, Ozer N, Ovünç K, Aksöyek S, Kes S.
Department of Cardiology, Hacettepe University School of Medicine, Ankara, Turkey.
Int J Cardiol. 2002 Aug;84(2-3):227-31.
The beneficial effects of HMG-CoA reductase inhibitors (statins) in patients with coronary artery disease (CAD) appear to be attributable not only to their lipid-lowering properties, but also
- to their therapeutic effects on the coagulation system, and anti-inflammatory effect.
Furthermore, statins mitigate the apoptosis of vascular smooth muscle cells (VSMC) and macrophages in atherosclerotic plaques.
The purpose of this study was to evaluate the effects of short-term atorvastatin treatment on
- the fibrinolytic system and systemic inflammatory status, and
- on apoptosis in hyperlipidemic patients with CAD.
The study population consisted of 36 hyperlipidemic patients (14 women and 22 men, mean age 53+/-9 years) with stable CAD, untreated with lipid-lowering medications. Serum lipoproteins, fibrinogen levels, sFas and sL-selectin, and global fibrinolytic capacity (GFC) were measured at baseline and after 12 weeks of treatment with atorvastatin, 10 mg/day.
Atorvastatin treatment decreased serum low-density lipoprotein (-39%, P=0.0001), total cholesterol (-32%, P=0.0001), and triglycerides (-22%, P=0.0001), and increased high-density lipoprotein (+13%, P=0.0001) at 12 weeks compared to baseline.
These effects were associated with
- a decrease in plasma fibrinogen from 331+/-73 to 298+/-58 mg/dl (P=0.0001), and
- sL-selectin levels from 666+/-201 to 584+/-162 ng/ml (P=0.0001).
- sFas levels increased from 3754+/-1264 to 4873+/-1835 pg/ml
- GFC from 3.5+/-2.4 to 5.6+/-2.9 microg/ml, respectively (both P=0.0001).
These results suggest that lipid lowering with atorvastatin therapy
- significantly increases GFC,
- decreases fibrinogen levels, and
- causes leukocyte deactivation.
Our findings also suggest that atorvastatin treatment mitigates apoptosis of VSMC in the atherosclerotic plaque. These effects of atorvastatin may, in part, explain the early decrease in cardiovascular events observed in clinical trials of statins. http://www.ncbi.nlm.nih.gov/pubmed/12127376PMID: 1212736
Atorvastatin induces associated reductions in platelet P-selectin, oxidized low-density lipoprotein, and interleukin-6 in patients with coronary artery diseases.
Oka H, Ikeda S, Koga S, Miyahara Y, Kohno S.
Second Department of Internal Medicine, School of Medicine, Nagasaki University, Nagasaki, Jp.
Abstract
The development and progression of atherosclerosis comprises various processes, such as endothelial dysfunction, chronic inflammation, thrombus formation, and lipid profile modification. Statins are 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors that have pleiotropic effects in addition to cholesterol-lowering properties. However, the mechanisms of these effects are not completely understood.
We investigated whether atorvastatin
- affects the levels of malondialdehyde-modified low-density lipoprotein (MDALDL), an oxidized LDL,
- the proinflammatory cytokine interleukin-6 (IL-6),
- platelet P-selectin, a marker of platelet activation, relative to that of LDL cholesterol (LDL-C).
Forty-eight patients with coronary artery disease and hyperlipidemia were separated into two groups that were administered with (atorvastatin group) or without (control group) atorvastatin. The baseline MDA-LDL level in all participants significantly correlated with LDL-C (r = 0.71, P < 0.01) and apolipoprotein B levels (r = 0.66, P < 0.01).
Atorvastatin (10 mg/day) significantly reduced the LDL-C level within 4 weeks and persisted for a further 8 weeks of administration. Atorvastatin also reduced the MDA-LDL level within 4 weeks and further reduced it over the next 8 weeks.
Platelet P-selectin expression did not change until 4 weeks of administration and then significantly decreased at 12 weeks, whereas the IL-6 level was gradually, but not significantly, reduced at 12 weeks. In contrast, none of these parameters significantly changed in the control group within these time frames.
The reduction (%) in IL-6 between 4 and 12 weeks after atorvastatin administration significantly correlated with that of MDALDL and of platelet P-selectin (r = 0.65, P < 0.05 and r = 0.70, P < 0.05, respectively). These results suggested that the positive effects of atorvastatin on the LDL-C oxidation, platelet activation and inflammation that are involved in atherosclerotic processes are exerted in concert after lowering LDL-C. http://www.ncbi.nlm.nih.gov/pubmed/18649055 PMID: 18649055
Simvastatin treatment improves endothelial function and increases fibrinolysis in patients with hypercholestrolemia.
Guven GS, Atalar E, Yavuz B, Beyazit Y, Kekilli M, Kilicarslan A, Sahiner L, Oz G, Ozer N, Aksoyek S, Haznedaroglu IC, Sozen T.
Hacettepe University Faculty of Medicine, Department of Internal Medicine, Ankara, Turkey
J Natl Med Assoc. 2006 Apr;98(4):627-30.
Statins reduce cardiovascular events by cholesterol-lowering as well as nonlipid-related actions. Thrombin activatable fibrinolysis inhibitor (TAFI) is a recently identified independent risk factor of thrombosis. The aim of this study was to assess the effects of simvastatin treatment on circulating TAFI concentrations and endothelial function in patients with hypercholesterolemia.
Treatment with simvastatin showed a significant decrement in plasma total cholesterol, LDL cholesterol and triglyceride levels (p<0.05). Plasma TAFI levels were also significantly decreased after simvastatin treatment [median 17.0 (range 0.4-93.7) mcg/mL versus median 6.9 (range 0.8-63.0) mcg/mL, p<0.001]. Mean FMD was measured 7.7 +/- 2.5% at baseline and significantly improved after treatment (13.0 +/- 1.4%) (p=0.001).
Our findings of decreased TAFI levels may reflect the beneficial effect of simvastatin treatment on fibrinolysis, and improved endothelial function may suggest the improved future cardiovascular events in hyperlipidemic patients.
http://www.ncbi.nlm.nih.gov/pubmed/16623076 PMID: 16623076 PMCID: PMC2569240
Endothelium dysfunction in the coronary circulation.
Lüscher TF, Noll G.
University Hospital, Bern, Switzerland.
Coronary artery disease pathogenesis involves an enhanced vasoconstriction and increased interaction of platelets and monocytes with the vessel wall, as well as proliferation, migration, and extracellular matrix formation of vascular smooth-muscle cells. The endothelium lies in a strategic anatomical position between the circulating blood and vascular smooth-muscle cells. This supports the concept that dysfunction of these cells plays a crucial role in the pathogenesis of coronary artery disease. Endothelial cells are a source of vasodilators such as
- nitric oxide,
- prostacyclin, and
- hyperpolarizing factor.
In addition, the cells produce heparin-like substances as well as other molecules with antiproliferative properties. These effects of endothelial cells may explain why
- platelets and monocytes usually do not adhere at the blood vessel wall and
- vascular smooth-muscle cells remain in a vasodilated state and neither migrate nor proliferate.
However, under pathological conditions, in particular in the presence of cardiovascular risk factors, endothelial dysfunction occurs and significantly
- contributes to the increase of platelet-vessel wall interaction, vasoconstriction, and proliferation in the coronary system.
Under these conditions, endothelium-dependent vasodilation is reduced and endothelium-dependent constrictor responses are augmented. Therapeutic strategies aimed at improving or preserving endothelial function therefore may be promising in the prevention and treatment of coronary artery disease. PMID: 7700060
Effect of Atherosclerosis on Endothelium-Dependent Inhibition of Platelet Activation in Humans
JG Diodati, N Dakak, DM Gilligan, AA Quyyumi
From the Cardiology Branch, National Heart, Lung, and Blood Institute (N.D., D.M.G., A.A.Q.), Bethesda, Md, and Cardiology Division, Department of Medicine, Jewish General Hospital, McGill University (J.G.D.), Montreal, Quebec, Canada
- Accepted Feb 25, 1998. Copyright © 1998 by American Heart Association
Circulation.1998; 98: 17-24doi: 10.1161/01.CIR.98.1.17
http://circ.ahajournals.org/content/98/1/17.full#ref-1
Subjects with risk factors or established atherosclerosis have depressed vasodilator responses to endothelium-dependent agonists, which are attributed largely to decreased abluminal availability of NO. Human studies examining the in vivo effects of luminally released NO on platelets are sparse, and it is unknown whether any inhibitory action of the vascular wall on platelets is attenuated in patients with endothelial dysfunction.
The vascular endothelium profoundly influences smooth muscle tone by the abluminal release of endothelium-derived relaxing factors. The predominant relaxing factor is nitric oxide (NO), a byproduct of L-arginine metabolism, which not only diffuses into the vascular smooth muscle layer but is also released luminally.
1HYPERLINK “http://circ.ahajournals.org/content/98/1/17.full#ref-2″2 3 4
In vitro, NO inhibits platelet adhesion and, to a lesser extent, platelet aggregation by increasing cytosolic levels of soluble cGMP. In whole blood, in which the half-life of NO is attenuated by hemoglobin and other oxidants, the platelet inhibitory effects of NO are less easily demonstrated and may be insignificant.
Stimulation of NO release into the vascular lumen with ACh inhibits platelet aggregation, an effect that is attenuated in patients with atherosclerosis and endothelial dysfunction.
- L-Arginine inhibited platelet aggregation by its direct action on platelets.
This reduced antiplatelet aggregatory property of the vasculature may predispose patients with atherosclerosis to thrombotic vascular events and, because of the known contribution of platelet-derived products to atherosclerosis, to more rapid progression of atherosclerosis.
These findings provide a pathophysiological basis for the observed increase in thrombotic events in atherosclerosis. L-Arginine supplementation and other therapies designed to improve endothelial NO activity should be tested for their long-term antithrombotic potential in atherosclerosis.
Furchgott RF, Zawadzki JV. The obligatory role of the endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. CrossRefMedline
Vanhoutte PM. The endothelium: modulator of vascular smooth muscle tone. N Engl J Med. 1988;319:512–513. Medline
Palmer R, Ferrige A, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. CrossRefMedline
Historical lessons in translational medicine: cyclooxygenase inhibition and P2Y12 antagonism.
Fitzgerald DJ, Fitzgerald GA.
UCD Conway Institute and Java Clinical Research, University College Dublin, Ireland.
Circ Res. 2013 Jan 4;112(1):174-94.
http://dx.doi.org/10.1161/CIRCRESAHA.111.300271
The development of drugs that inhibit platelets has been driven by a combination of clinical insights, fundamental science, and sheer luck. The process has evolved as the days of stumbling on therapeutic gems, such as aspirin, have long passed and have been replaced by an arduous process in which a drug is designed to target a specific protein implicated in a well-characterized pathophysiological process, or so we would like to believe. The development of antiplatelet therapy illustrates the importance of understanding the mechanisms of disease and the pharmacology of the compounds we develop, coupled with careful clinical experimentation and observation and, yes, still, a fair bit of luck. http://www.ncbi.nlm.nih.gov/pubmed/23287454
PMID: 23287454 PMCID: PMC3572712 [Available on 2014/1/4]
The origin and function of platelet glycosyltransferases
HH Wandall, V Rumjantseva, AL Tølbøll Sørensen, S Patel-Hett, EC Josefsson, et al.
1 Center for Glycomics, Cell Molec Med, Health Sciences, Univ of Copenhagen, Copenhagen, Den; and
2Translational Medicine Division, Depart Med, B&W Hosp, Harvard Med School, Boston, MA
Platelets are megakaryocyte subfragments that participate in hemostatic and host defense reactions and deliver pro- and antiangiogenic factors throughout the vascular system. Although they are anucleated cells that lack a complex secretory apparatus with distinct Golgi/endoplasmic reticulum compartments, past studies have shown that platelets have glycosyltransferase activities.
We show that members of 3 distinct glycosyltransferase families are found within and on the surface of platelets. Immunocytology and flow cytometry results indicated that megakaryocytes package these Golgi-derived glycosyltransferases into vesicles that are sent via proplatelets to nascent platelets, where they accumulate. These glycosyltransferases are active, and intact platelets glycosylate large exogenous substrates.
We also show that
- activation of platelets results in the release of soluble glycosyltransferase activities and
- platelets contain sufficient levels of sugar nucleotides for detection of glycosylation of exogenously added substrates.
Therefore, the results of the present study show that blood platelets are a rich source of both glycosyltransferases and donor sugar substrates that can be released to function in the extracellular space. This platelet-glycosylation machinery offers a pathway to a simple glycoengineering strategy improving storage of platelets and may serve hitherto unknown biologic functions.
http://bloodjournal.hematologylibrary.org/content/120/3/626.abstract
Accepted May 14, 2012. © 2012 by The American Society of Hematology
Systems biology of platelet-vessel wall interactions
Scott L. Diamond*, Jeremy Purvis, Manash Chatterjee and Matthew H. Flamm
Department of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA
Fronteirs in Physiology Aug 2013; 4(229). http://dx.doi.org/10.3389/fphys.2013.00229
Blood systems biology seeks to quantify outside-in signaling as platelets respond to numerous external stimuli, typically under flow conditions. Platelets can activate via GPVI collagen receptor and numerous G-protein coupled receptors (GPCRs) responsive to ADP, thromboxane, thrombin, and prostacyclin. A bottom-up ODE approach allowed prediction of platelet calcium and phosphoinositides following P2Y1 activation with ADP, either for a population average or single cell stochastic behavior. The homeostasis assumption (i.e., a resting platelet stays resting until activated) was particularly useful in finding global steady states for these large metabolic networks. Alternatively, a top-down approach involving Pairwise Agonist Scanning (PAS) allowed large data sets of measured calcium mobilization to predict an individual’s platelet responses. The data was used to train neural network (NN) models of signaling to predict patient-specific responses to combinatorial stimulation. A kinetic description of platelet signaling then allows prediction of inside-out activation of platelets as they experience the complex biochemical milieu at the site of thrombosis. Multiscale lattice kinetic Monte Carlo (LKMC) utilizes these detailed descriptions of platelet signaling under flow conditions where released soluble species are solved by finite element method and the flow field around the growing thrombus is updated using computational fluid dynamics or lattice Boltzmann method. Since hemodynamic effects are included in a multiscale approach, thrombosis can then be predicted under arterial and venous thrombotic conditions for various anatomical geometries. Such systems biology approaches accommodate the effect of anti-platelet pharmacological intervention where COX1 pathways or ADP signaling are modulated in a patient-specific manner.
Keywords: platelet, thrombosis, hemodynamic, ADP, thromboxane
A systems biology approach for platelets seeks to predict kinetic processes during clotting or bleeding episodes. A mul-tiscale modeling framework should facilitate the bridging of genomics/proteomics studies with platelet phenotype and vessel pathophysiology under hemodynamic conditions. Such a framework should quantify risks and severity of such episodes for a given phenotype/genotype as well as the safety and efficacy of pharmacological intervention.
Genome-wide association studies (GWAS) have found rather modest associations of SNPs (single nucleotide polymorphisms) with thrombosis or vascular disease (Wellcome Trust Case Control Consortium, 2007; Ouwehand, 2007). A GWAS focused specifically on coronary artery disease (CAD) identified 4 loci linked to CAD (Samani et al., 2007), but mechanistic understanding of these SNPs awaits exploration and may not necessarily be platelet-linked. Jones et al. (2007) measured platelet response, calcium mobilization, aggregometry, and FACS determination of response to ADP or GPVI activation with collagen-related peptide (CRP) in 506 healthy volunteers to define inter-individual vari-ability. Importantly, SNPs in the GP6 locus were linked to about 35% of the variation in response to CRP. Variability in response to ADP was associated with polymorphisms in the platelet P2Y12 receptor (Fontana et al., 2003). However, it currently remains difficult to use GWAS or genotyping information to predict risk for a specific patient. Greliche et al. (2013) conclude from their genome-wide SNP interaction analysis that common SNPs were unlikely to have strong interactive impact on the risk of venous thrombosis. Few if any genomic studies quantitatively predict an individual’s blood function during clotting or bleeding disease scenarios under hemodynamic conditions.
Platelets contain a microRNA (miR) pool and a translatable mRNA pool that declines with a platelet’s age in the circulation. McRedmond et al. (2004) identified 2928 mRNA species using Affymetrix arrays. Many of the top 50 most abundant platelet mRNAs correlated with secreted or membrane proteins such as β2-microglobulin, platelet factor 4, factor XIII, GPIb, αIIb, etc. Similarly, Gnatenko et al. (2003) found 2147 mRNA species and Bugert et al. (2003) found 1526 mRNA species in purified platelets. More recent direct sequencing methods identified ∼9000 mRNA species in platelets (Bray et al., 2010), more comparable in size to the transcriptome of megakaryocytes. Also, platelets contain a functional spliceosome. For example, the processing of tissue factor (TF) mRNA and translation of TF upon platelet activation was found in activated platelets (Schwertz et al., 2006), although TF activity was only detectable in sonicated platelet membranes. The human platelet proteome has been examined and an important web-based resource is now available to explore protein-protein interactions within platelets (Boyanova et al., 2011).
Inter-individual variation in platelet reactivity, even in a normal population, has been associated with a number of factors including: female gender, fibrinogen level, ethnicity, inherited variations, and polymorphisms (Hetherington et al., 2005; Bray, 2007). Unfortunately, no single genomic or proteomic factor is a strong predictor of hyper-reactivity in typical subjects and the need for advanced functional phenotyping motivates the development of systems biology tools to quantify blood function.

FIGURE 1 | Detailed reaction schemes for platelet signaling modules. Four interconnected models were defined: (A) Ca2+ module: cytosolic and DTS compartments are separated by the DTS membrane, which contains the IP3R and SERCA. (B) Phosphoinositide (PI) module: Membrane-bound PIs are cleaved by PLC-to form diffusible inositol phosphates and DAG, which are substrates for resynthesis of PIs. (C) PKC module: Ca2+i and DAG activate PKC, which migrates to the plasmamembrane where it phosphorylates PLC-. (D) P2Y1 module: extracellular ADP binds to and activates P2Y1. Active P2Y1 accelerates guanine nucleotide exchange on bound Gq. The Gq·GTP binds and activates PLC-, which increases the GTPase activity of Gq·GTP.
Molecular kinetic descriptions are embedded in the signaling modules for: (E) SERCA catalytic cycle (Dode et al., 2002): Subscripts: E1, facing cytosol; E2, facing DTS; P, phosphorylated. (F) IP3R dynamics (Sneyd and Dufour, 2002): Subscripts: n, native; i1, inhibited; o, open; a, active; s, shut, i2, inhibited. (G) PKC activation: Active kinase is bound to Ca2+i and DAG and located at the PM. Subscripts: M, located at the
PM; C, Ca2+-bound; D, DAG-bound. (H) P2Y1 activation module: Rate equations describing the interactions among ADP, P2Y1, and Gq were modeled according to the ternary complex model described in Kinzer-Ursem and Linderman (2007). Abbreviations: A, ADP; P, P2Y1; G, Gq. ∗Ca2+ both activates and inhibits IP3R
For multi-scale modeling of platelet-vessel wall interactions, a given modeling route at each scale has advantages and disadvantages. Top-down models (like NNs) are most easily obtained in a patient-specific manner to describe platelet function, however they typically fail to identify specific intracellular regulators. Bottom-up models (like ODE models) of platelet signaling provide molecular precision but face three challenges: (1) difficulty in parameterization, (2) difficulty in fitting to high dimensional data, and (3) incomplete knowledge. Both NN and ODE models are both relatively fast in terms of computational speed. Lattice kinetic Monte Carlo (LKMC) methods provide a balance of speed and sub-micron spatial precision, particularly for discrete cellular systems over millimeter-scale phenomenon and 100 or 1000s of cells. LKMC methods are also relatively easy to program and facilitate the passing of information with other lattice based methods (like Lattice Boltzman or finite elements). LKMC methods become exceedingly slow for molecular simulations of large ensembles when time steps become impossibly small. For solving 2D flow problems, Lattice Boltzman is relatively fast and easy to implement and has no issues of numerical stability. One of the biggest numerical challenges is solving multi-component, reaction-diffusion problems with spatial gradients. Wall-derived TF triggers coagulation and ∼10–100 PDEs must be solved to calculate thrombin and fibrin levels in a growing thrombus. Solving large PDE systems is especially computationally intensive (days or weeks) and resists parallel processing.
A central goal in blood systems biology is to elucidate the regulatory complexity of cellular signaling across a large ensemble of interacting cells responding to numerous spatiotemporal stimuli in the presence of pharmacological mediators, ideally in a patient-specific and disease-relevant context (i.e., containing hemodynamics). Developing tools to define platelet variations between patients and the relationship of platelet phenotype to prothrombotic or bleeding traits will have significant impact in stratifying patients according to risk. This multiscale approach also makes feasible patient-specific prediction of platelet deposition and drug response in more complex in vivo geometries such as stenosis, aneurysms, stented vessels, valves, bifurcations, or vessel rupture (for prediction of bleeding risks) or in geometries encountered in mechanical biomedical devices.
However, it currently remains difficult to use GWAS or genotyping information to predict risk for a specific patient. Greliche et al. (2013) conclude from their genome-wide SNP interaction analysis that common SNPs were unlikely to have strong interactive impact on the risk of venous thrombosis. Few if any genomic studies quantitatively predict an individual’s blood function during clotting or bleeding disease scenarios under hemodynamic conditions.
Platelets contain a microRNA (miR) pool and a translatable mRNA pool that declines with a platelet’s age in the circulation. McRedmond et al. (2004) identified 2928 mRNA species using Affymetrix arrays. Many of the top 50 most abundant platelet mRNAs correlated with secreted or membrane proteins such as β2-microglobulin, platelet factor 4, factor XIII, GPIb, αIIb, etc. Similarly, Gnatenko et al. (2003) found 2147 mRNA species and Bugert et al. (2003) found 1526 mRNA species in purified platelets. More recent direct sequencing methods identified ∼9000 mRNA species in platelets (Bray et al., 2010), more comparable in size to the transcriptome of megakaryocytes. Also, platelets contain a functional spliceosome. For example, the processing of tissue factor (TF) mRNA and translation of TF upon platelet activation was found in activated platelets (Schwertz et al., 2006), although TF activity was only detectable.
Scientists Discover Novel Functions of Platelets
Sep. 1, 2013 — A new finding could lead to novel treatments to reduce bleeding in trauma and severe infections.
The research, from Oklahoma Medical Research Foundation scientists Lijun Xia, M.D., Ph.D., Jianxin Fu, M.D., Ph.D., and Brett Herzog, Ph.D., appears in the most recent issue of the journal Nature.
Xia and his team of researchers found that platelets, which normally stop blood loss by clumping and forming plugs in blood vessel holes after injuries, activate a screening process. And this process allows lymphocytes to exit into lymph nodes without letting red blood cells leave the blood vessel.
“Platelets are the smallest blood cells that work in clotting to heal cuts because they stick to the site of the injury,” said Xia, a member of OMRF’s Cardiovascular Biology Research Program. “This novel function requires platelets to dump a specific lipid content, but does not need intact platelets because it’s not forming a clot. We never knew they could do this before.”
Not only are platelets making it possible for lymphocytes to leave the blood vessel, they’re doing so by going outside the vessel, themselves — another novel finding, he said.
When scientists interrupted the process by removing a protein called podoplanin, the screening process stopped working, allowing both lymphocytes and red blood cells to escape. The new study reveals a novel function of platelets independent of their hemostatic role. The findings could alter the ways in which doctors use platelets to treat traumatic injuries and serious infections.
Intact platelets that can clot usually only last 5 to 7 days in the blood and cannot be frozen, making storage a problem, Xia said. Since these new functions do not rely on intact platelets, it points to different uses for platelets, perhaps even some that have been frozen. If it works out, the discovery could be useful in stopping widespread internal bleeding caused by explosive traumas or severe infections.
“As the research continues, I think there’s a possibility this will lead to new therapeutics that could slow or stop hemorrhaging in trauma and sepsis-related illnesses,” Xia said.
The breakthrough has opened several avenues for further research, he said. One will be a better understanding of how platelets go outside the vessels to start the process. They will also be looking to see if platelets perform the same role in letting rapid-attack immune cells called neutrophils out of the bloodstream to fight infections.
“This is a prime example of the important research that the Institutional Development Award program makes possible in states that have historically had low levels of funding from the National Institutes of Health,” said NIH grant program official María Teresa Canto, D.D.S., M.S., M.P.H. “Dr. Xia’s study sheds light on a process that is key to vascular health as well as to the development of inflammation and associated diseases.”
Story Source: The above story is based on materials provided by Oklahoma Medical Research Foundation, via EurekAlert!, a service of AAAS.
Journal Reference:
Podoplanin maintains high endothelial venule integrity by interacting with platelet CLEC-2.
BH Herzog, J Fu, SJ Wilson, PR Hess, A Sen, JM McDaniel, Y Pan, M Sheng, et al. Nature, 2013; http://dx.doi.org/10.1038/nature12501
Oklahoma Medical Research Foundation (2013, September 1). Scientists discover novel functions of platelets. ScienceDaily. Retrieved September 27, 2013, from
http://www.sciencedaily.com /releases/2013/09/130901154022.htm
Synthetic Blood Platelets Developed
May 30, 2012 — Synthetic platelets have been developed by UC Santa Barbara researchers, in collaboration with researchers at Scripps Research Institute and Sanford-Burnham Institute in La Jolla, Calif. Their findings are published in the journal Advanced Materials.
The synthetic platelets represent the latest and one of the most advanced in a line of efforts over the last century to mimic platelet function. While clotting factors and platelets from outside donors are used widely to halt bleeding, immune system responses and thrombosis have been issues.
“This development is a significant milestone in the field of biomimetic materials,” said Samir Mitragotri, professor of chemical engineering, director of UC Santa Barbara’s Center for Bioengineering, and an author of the paper. “By capitalizing on our capabilities in engineering materials, with the expertise in platelet biology that exists in Professor Ruggeri’s laboratory, our synthetic platelets combine unique physical and biological attributes that mimic natural platelets.
Other authors of the study include Jennifer N. Orje, Blanca Molins, and Zaverio Ruggeri from Scripps Research Institute; and Jeffrey Smith from Sanford-Burnham Medical Research Institute. This work was funded by the Institute for Collaborative Biotechnologies through a grant from the U.S. Army Research Office and the National Institutes of Health.
http://www.sciencedaily.com/releases/2012/05/120530133738.htm
Synthetic Platelets Halve Clotting Time
In an effort to enhance the natural process, a team led by Erin Lavik, a new Case Western Reserve University biomedical engineering professor, and her former doctoral student, James P. Bertram, built synthetic platelets that show promise in halting internal and external bleeding.
Their work is published in Science Translational Medicine.
The researchers were inspired by studies showing there are few options to treat soldiers suffering from internal injuries in Afghanistan and Iraq. They wanted to develop a treatment medics can keep in their field packs.
“The military has been phenomenal at developing technology to halt bleeding, but the technology has been effective only on external or compressible injuries,” Lavik said. “This could be a compliment to current therapies.”
Blood platelets are the structural and chemical foundation of blood clotting, a complex cascade of events that works well with normal cuts and scrapes but can be overmatched by serious injury.
Using other’s platelets can enhance clotting but carries risks of several complications. And these platelets must be refrigerated and have a short shelf life.
Bertram and Lavik developed platelets made from biodegradable polymers. The synthetic platelets are designed to home in and link up with natural platelets at the site of an injury.
In essence, adding artificial platelets to a traumatic injury site is akin to adding sand bags to a levy along a flooding river.
The natural platelets, activated by injury, emit chemicals that bind natural platelets and the additional synthetics into a larger clot that quickly stems the bleeding.
http://www.sciencedaily.com/releases/2009/12/091216144139.htm
Novel Antiplatelet Therapy
Platelet Biology—Focus on P2Y12 and Thrombin Receptors
MA Gaglia Jr., SV Manoukian, R Waksman
American Heart Journal. 2010;160(4):595-604. http://www.medscape.com/viewarticle/730897_2
An understanding of the role of platelets in primary hemostasis is necessary to comprehend the process of thrombosis that ultimately results in ACS. Although a complete review of this intricate process is beyond the scope of this review, an overview with particular attention to the importance of the platelet agonists adenosine diphosphate (ADP) and thrombin follows.
Primary hemostasis is composed of three essential phases:
-
platelet adhesion,
-
activation, and
-
aggregation.
Disruption of the endothelium, through either vessel injury or atherosclerotic plaque rupture, exposes tissue factor; this activates a pathway that ultimately liberates thrombin through the coagulation pathway. The extracellular matrix is also exposed, thus initiating platelet adhesion.
The most important substrates for platelet binding are collagen, which binds the glycoprotein (GP) Ib/IX/V platelet receptor, and von Willebrand Factor, which binds the GP VI receptor. Adhesion initiates platelet activation through two key processes:
-
shape change of the platelet surface membrane, which results in membrane protrusions to facilitate tethering to other platelets; and
-
the activation of complex intracellular signaling pathways that ultimately result in the release of platelet agonists such as ADP, thromboxane A2, and thrombin.
Such agonists subsequently activate other platelets. The last step in this process, aggregation, is facilitated by the expression of GpIIb-IIIa on the surface of activated platelets. This receptor undergoes a shape change with activation, allowing it to bind fibrinogen and von Willebrand Factor, thereby effecting the cross-linking of platelet to platelet and ultimately leading to the formation of a platelet thrombus.
Receptors on the platelet surface are vital to the above process. This includes the thromboxane A2 receptor (which has two subtypes, TPα and TPβ), the GpIIb-IIIa receptor, the P2Y12 receptor, and the protease activated receptors (PAR). The P2Y12 receptor is coupled to the Gi protein and is activated by the binding of ADP, which then liberates the αi and βγ subunits. The αi subunit inhibits adenylyl cyclase, resulting in a decrease in intracellular cyclic adenosine monophosphate (cAMP) levels. This thereby decreases cAMP-mediated phosphorylation of the vasodilator-stimulated phosphoprotein (VASP).This protein is important for two reasons: first, it results in activation of the GpIIb-IIIa receptor; second, measurement of its phosphorylated form (VASP-P) is essentially a measure of overall P2Y12 activity, with higher levels of VASP-P representing relatively more P2Y12 inhibition. This has been applied with good effect to the VASP phosphorylation assay, which uses flow cytometry to measure levels of VASP-P. The βγ subunit of the Gi protein has a similar effect, but through a different mechanism. βγ activates phosphatidylinositol-3-kinase, which then activates both a serine threonine protein kinase B and Rap1b. These guanosine 5′-triphosphate-binding proteins then lead to GpIIb-IIIa receptor activation.
As our understanding of platelet biology grows, the PAR-mediated pathway is also emerging as a therapeutic target. Human platelets express two types of PAR, PAR-1 and PAR-4. Both are G-protein coupled receptors that bind thrombin, the most potent known physiological agonist of platelets that also is crucial to the coagulation cascade. PAR-1 appears to have higher affinity for thrombin than PAR-4, although the relative importance of PAR-1 versus PAR-4 is still somewhat unclear. Thrombin binding to PAR-1 cleaves the receptor, initiating an intracellular signaling process facilitated by various G-proteins. Specifically, G12 and G13 ultimately lead to cytoskeletal changes that likely mediate platelet shape change; whereas Gq and Gi lead to increased intracellular cAMP and calcium, leading to GpIIb-IIIa activation and thereby contributing to platelet activation.
Endothelial and Vascular Smooth Cell Dysfunctions: A Comprehensive Appraisal
Luigi Fabrizio Rodella and Rita Rezzani
Department of Biomedical Sciences and Biotechnology, University of Brescia, Italy
Cardiovascular disease (CvDs) such as coronary artery disease, hypertension, congestive heart failure and stroke are the leading causes of death and disability in the Western World (Madamanchi et al., 2005; Thom, 1989). The majority of CvDs results from complication of atherosclerosis. Prevention of cardiovascular events is therefore urgently needed and is one of the major recent challenges of medicine. New molecular imaging approaches featuring the assessment of inflammatory processes in the vascular wall (on top of existing anatomic and functional vessel imaging procedures) could emerge as decisive tools for the understanding and prevention of cardiovascular events (Schafers et al., 2010).
Atherosclerosis is a progressive disease, affecting medium and large-sized arteries, characterized by patchy intramural thickening of the subintimal that encroaches on the arterial lumen (Bonomini et al., 2008). The atherosclerosis plaque is characterized by an accumulation of lipid in the artery wall, together with infiltration of macrophages, T cells and mast cells, and the formation by vascular smooth muscle cells (VSMCs) of a fibrous cap composed mostly of collagen. Early lesions called “fatty streaks” consist of sub-endothelial deposition of lipid, macrophage foam cells loaded with cholesterol and T cells. Over time, a more complex lesion develops, with apoptotic as well as necrotic cells, cell debris and cholesterol crystals forming a necrotic core in the lesion. This structure is covered by a fibrous cap of variable thickness, and its “shoulder” regions are infiltrated by activated T cells, macrophages and mast cells, which produce proinflammatory mediators and enzymes (Hansson et al., 2006). Plaque growth can cause stenosis (narrowing of the lumen) that can contribute to ischemia in the surrounding tissue (Hansson & Hermansson, 2011). Although the pathophysiological mechanisms underlying atherosclerosis are not completely understood, it is widely recognized that both inflammation and oxidative stress play important roles in all of the phases of atherosclerosis evolution (Cipollone et al., 2007).

Fig. 1. Steps involved in atherosclerosis progression from endothelial dysfunction to cardiovascular complication
Oxidative stress can be defined as an “imbalance between oxidants and antioxidants in favor of the oxidants, potentially leading to damage” (Sies, 1991). Age, gender, obesity, cigarette smoking, hypertension, diabetes mellitus and dyslipidemia are known atherogenic risk factors that promote the impairment of endothelial function, smooth muscle function and vessel wall metabolism. These risk factors are associated with an increased production of reactive oxygen species (ROS) (Antoniades et al., 2003). ROS play a physiological role in the vessel wall and participate as second messengers in endothelium-dependent function, in smooth muscle cells and endothelial cells (ECs) growth and survival, and in remodelling of the vessel wall. Each of these responses, when uncontrolled, contributes to vascular diseases (Fortuño et al., 2005; Griendling & Harrison, 1999; Irani, 2000; Taniyama & Griendling, 2003).
In the vasculature wall, ROS are produced by all the layers, including tunica intima, media and adventitia. ROS include superoxide anion radical (O2–), hydrogen peroxide (H2O2), hydroxyl radical (OH), nitric oxide (NO), and peroxynitrite (ONOO–) (Lakshmi et al., 2009). The major vascular ROS is O2−, which inactivates NO, the main vascular relaxing factor, thus impairing relaxation (Cai & Harrison, 2000; Kojda & Harrison, 1999). Dismutation of O2− by superoxide dismutase (SOD) produces H2O2, a more stable ROS, which, in turn, is converted to water by catalase and glutathione peroxidase. H2O2 and other peroxides appear to be important in the regulation of growth-related signalling in VSMCs and inflammatory responses in vascular lesions (Irani, 2000; Li, P.F. et al., 1997). High levels of O2−, the consequent accumulation of H2O2 and diminished NO bioavailability play a critical role in the modulation of vascular remodelling. Finally, ONOO–, resulting from the reaction between O2− and NO, constitutes a strong oxidant molecule, which is able to oxidize proteins, lipids and nucleic acids and then causes cell damage (Beckman & Koppenol, 1996; Fortuño et al., 2005).
There are several potential sources of ROS production. In cardiovascular disease the sources include xanthine oxidase, cyclooxygenase, lipooxygenase, mitochondrial respiration, cytochrome P450, uncoupled nitric oxide synthase (NOS) and NAD(P)H oxidase. They have been identified as sources of ROS generation in all type of vasculature. These sources may contribute to ROS formation, depending on cell type, cellular activation site and disease context. Numerous studies have shown that various physiological stimuli that contribute to pathogenesis of vascular disease can induce the formation of ROS (Lakshmi et al., 2009). ROS have detrimental effects on vascular function through several mechanisms. First, ROS, especially hydroxyl radicals, directly injure cell membranes and nuclei. Second, by interacting with endogenous vasoactive mediators formed in ECs, ROS modulate vasomotion and the atherogenic process. Third, ROS peroxidize lipid components, leading to the formation of oxidized lipoproteins (LDL), one of the key mediators of atherosclerosis (Bonomini et al., 2008).
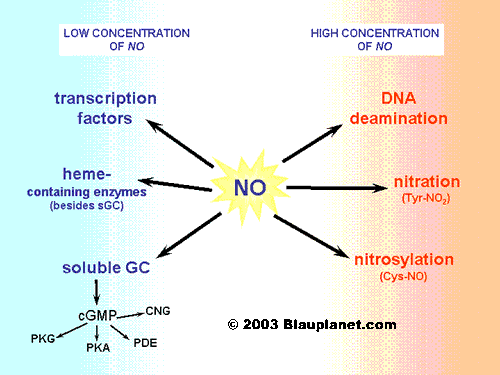

Fig. 2. Potential sources of ROS production in atherosclerosis progression.
Cholesterol is transported in the blood by LDL. These particles contain esterified cholesterol and triglycerides surrounded by a shell of phospholipids, free cholesterol and apolipoprotein B100 (ApoB100). Circulating LDL particles can accumulate in the intimal, the innermost layer of the artery. Here ApoB100 binds to proteoglycans of the extracellular matrix (ECM) through ionic interactions (Tabas et al., 2007). This is an important initiating factor in early atherogenesis (Skålen et al., 2002; Steinberg, 2009; Witztum & Steinberg, 2001). As a consequence of this subendothelial retention, LDL particles are trapped in the tunica intima, where they are prone to oxidative modifications caused by enzymatic attack of myeloperoxidase (Heinecke, 2007) and lipoxygenases, or by ROS such as hypoclorous acid (HOCl), phenoxyl radical intermediates or ONOO– generated in the intimal during inflammation and atherosclerosis (Hansson & Hermansson, 2011).
Oxidized LDL (Ox-LDL) has several biological effects (Madamanchi et al., 2005); it is pro-inflammatory; it causes inhibition of endothelial NOS (eNOS); it promotes vasoconstriction and adhesion; it stimulates cytokines such as interleukins (ILs) and increases platelet aggregation. Ox-LDL-derived products are cytotoxic and induce apoptosis. Ox-LDL can adversely affect coagulation by stimulating tissue factor and plasminogen activator inhibitor-1 (PAI-1) synthesis. Another atherogenic property of Ox-LDL is its immunogenicity and its ability to promote retention of macrophages in the arterial wall by inhibiting macrophage motility (Singh & Jialal, 2006). In addiction, Ox-LDL stimulates VSMCs proliferation (Stocker & Keaney, 2004). Thus, intimal thickening further reduces the lumen of blood vessels, leading to further potentation of hypertension and atherosclerosis (Singh & Jialal, 2006). With ongoing oxidation, the physicochemical properties gradually change, including alterations in charge, particle size, lipid content and other features. The precise nature of each of these alterations obviously depends on the oxidizing agent. For all these reasons, Ox-LDL is not a defined molecular species but is instead a spectrum of LDL particles that have undergone a variety of physicochemical changes (Hansson & Hermansson, 2011).
Inflammation participates in atherosclerosis from its inception onwards. Fatty streaks do not cause symptoms, and may either progress to more complex lesions or involute. Fatty streaks have focal increases in the content of lipoproteins within regions of the intimal, where they associate with components of the ECM such as proteoglycans, slowing their egress. This retention sequesters lipoproteins within the intimal, isolating them from plasma antioxidants, thus favoring their oxidative modification (Kruth, 2002; Packard & Libby, 2008; Skålen et al., 2002). Oxidatively modified LDL particles comprise an incompletely defined mixture, because both the lipid and protein moieties can undergo oxidative modification. Constituents of such modified lipoprotein particles can induce a local inflammatory response (Miller et al., 2003; Packard & Libby, 2008).
Vascular ECs function to prevent clotting of blood and adhesion of blood cells to the endothelial cells, in addition to playing the role of a barrier, as a cell monolayer, to prevent blood constituents from invading the vascular wall. When ECs are injured or activated by various coronary risk factors, infections or physical stimuli, adhesion molecules become expressed in ECs, and peripheral monocytes adhere to the endothelial cell surface. Adhesion
molecules are broadly divided into three molecular families:
-
integrin family,
-
immunoglobulin family, and
-
selectin family (L-selectin, Eselectin, P-selectin) (Yamada, 2001).
Chemoattractant factors, which include monocyte chemoattractant protein-1 (MCP-1) produced by vascular wall cells in response to modified lipoproteins, direct the migration and diapedesis of adherent monocytes (Boring et al., 1998; Packard & Libby, 2008). Monocytic cells, directly interacting with human ECs, increase several fold monocyte matrix metalloproteinase (MMP) 9 production, allowing for the subsequent infiltration of leukocytes through the endothelial layer and its associated basement membrane (Amorino & Hoover, 1998; Packard & Libby, 2008) Within the intima, monocytes mature into macrophages under the influence of macrophage colony stimulating factor (M-CSF), which is overexpressed in the inflamed intima. M-CSF stimulation also increases macrophage expression of scavenger receptors, members of the pattern-recognition receptor superfamily, which engulf modified lipoproteins through receptor-mediated endocytosis. Accumulation of cholesteryl esters in the cytoplasm converts macrophages into foam cells, i.e., lipid-laden macrophages characteristic of early-stage atherosclerosis. In parallel, macrophages proliferate and amplify the inflammatory response through the secretion of numerous growth factors and cytokines, including tumor necrosis factor α (TNFα) and IL-1β. Recent evidence supports selective recruitment of a proinflammatory subset of monocytes to nascent atheroma in mice (Packard & Libby, 2008).
A number of proinflammatory cytokines have been shown to participate in atherosclerotic plaque development, growth and rupture (Dabek, 2010; Libby et al., 2002). Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) seems to be a crucial transcription factor in the cross-talk among cytokines, adhesion molecules and growth factors. On one hand, NF-kB is a major transcription factor leading to cytokine synthesis, and on the other hand, the above mentioned factors keep NF-kB persistently activated in acute coronary syndromes (Dabek, 2010). In atherogenesis, NF-kB before regulates the expression of cyclooxygenases, lipooxygenases, cytokines, chemokines (i.e., MCP-1) and adhesion molecules (Dabek, 2010; Kutuk & Basaga, 2003). Later in the progression of the atherosclerotic lesion, NF-kB regulates gene expression of M-CSF, a factor stimulating infiltrating monocyte differentiation and transformation into “foamy cells”, and other genes participating in the transformation (Brach et al., 1991; Dabek, 2010). As stated, atherosclerosis is an inflammatory reaction of the arterial wall. The factors IL-1β, TNF-α, IL-6, IL-12 and interferon γ (IFNγ) are involved in this reaction and their expression is coregulated by NF-kB.
Intracellular matrix degradation is an important process in both plaque development and rupture. The vital factors involved include MMPs, particularly those that are able to break down the vascular base membrane. It has been shown that NF-kB is an essential regulator of MMP gene expression, especially MMP-2 and MMP-9, which are critical in plaque rupture (Bond et al., 1998; Dabek, 2010). Thus, NF-kB regulates the expression of a wide spectrum of atherosclerosis mediating factors. On the other hand, most of these factors also up-regulate NF-kB activity. Increased NF-kB activity was found in unstable regions of atherosclerotic plaques (Brand et al., 1997; Dabek, 2010). The significance of NF-kB activity has been confirmed in some clinical studies as well. Li and colleagues reported significantly increased NF-kB activity in white blood cells from unstable angina patients vs. stable angina patients and vs. control patients (the lowest activity in the latter) (Li, J.J. et al., 2004).


Fig. 4. Central role of ROS in inducing endothelial dysfunction in vascular diseases.
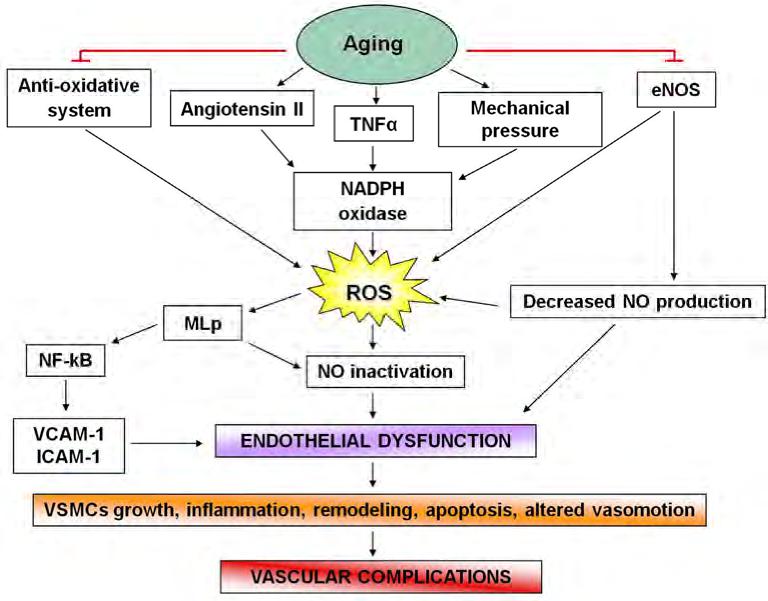

Fig. 8. Involvement of VSMCs apoptosis in fibrous plaque rupture.
The contemporary paradigm explaining smooth muscle replication in the vessel wall is that dysfunctional endothelium and/or inflammatory cells produce growth factors and ECM proteins that can induce replication and migration of VSMCs from the media to the intima (van Oostrom et al., 2009). In his “response-to injury” hypothesis, Ross proposed that VSMCs in the wall normally exist in a quiescent state, but, when the endothelium is injured, platelets release factors that stimulate VSMCs movement into and replication within the arterial intima (Ross, 1981-1982).
Growth factors have been known to influence the differentiated state of VSMCs (Willis et al., 2004). An interesting possibility is that smooth muscle replication may be controlled by factors intrinsic to the vessel wall. One possibility comes from evidence that normal endothelium contains inhibitors of smooth muscle proliferation (Haudenschild & Schwartz, 1979). The principal factor involved in VSMCs replication is the platelet derived growth factor (PDGF), which is a potent VSMC mitogen linked to vascular homeostasis and atherogenesis (Majesky et al., 1992). This peptide not only is mitogenic for VSMCs, but is chemotactic as well (Schwartz et al., 1986): the data on PDGF and its receptor subunits suggest, infact, a role in migration/localization of primordial VSMCs to the endothelium. This growth factor consists of two chain types, A and B, giving rise to three different PDGF subtypes (AA, AB, BB): PDGF-BB and -AB are known VMSCs chemoattractans, whereas PDGF-AA is associated with inhibition of chemotaxis (Zachary et al., 1999). PDGF binds to specific dimeric receptors (α and 3) found on smooth muscle cells (Bowen-Pope & Ross, 1982) where initiates a series of events leading to DNA synthesis: receptor α can bind all PDGF subtypes, while receptor 3 binds only subtypes -AB and -BB. VSMCs have been determined to upregulate expression of receptor 3 in response to vascular injury, inducing their chemotaxis; at the same time, these cells are able to increase the PDGF-AA, acting as a paracrine or autocrine regulator of their chemotaxis. This represents the first described autoregulation pathway of VSMCs on their own proliferation/migration (Willis et al., 2004). The second known requirement for cell cycle progression is availability of insulin-like growth factor (IGF-1), a co-factor that VSMCs require for completion of the cell cycle following stimulation with PDGF (Clemmons, 1984). Perhaps more surprising is that, as reported above, VSMCs may be able to stimulate their own growth by synthesis of both PDGF and IGF-1 (PDGF is able to stimulate smooth muscle cells to produce IGF-1).
Atherosclerosis and its associated complications remain the primary cause of death of the 21st century in humans. Recently it has been suggested that atherosclerosis is a multifactorial, multistep disease. Clinical and histopathological studies of atherosclerotic patient groups have identified inflammatory and oxidative stress-linked mechanisms as being pathogenetically important in atherosclerosis at every step from initiation to progression. Endothelial damage is also crucial for the progress of atherosclerosis and risk factors for atherosclerosis represent crucial factors associated with endothelial dysfunction. Studies have shown that patients with cardiovascular disease are characterized by impaired endothelial function, being vascular endothelium responsible for the secretion of several substances exerting proved anti-atherogenic effects. Finally, VSMCs are an important component of atherosclerotic plaques, responsible for promoting plaque stability in advanced lesions. In contrast, VSMC apoptosis has been implicated in a number of deleterious consequences of atherosclerosis, including plaque rupture, vessel remodelling, coagulation, inflammation and calcification. A better understanding of the pathogenesis of atherosclerosis will aid in for reducing mortality. An in-depth knowledge of the various pathogenic mechanisms involved in atherosclerosis can help in formulating preventive and therapeutic strategies and devising pharmaceutical and lifestyle modifications for reducing mortality.
Pharmacodynamic Properties of Antiplatelet Agents
Current Knowledge and Future Perspectives
KI Kalantzi, ME Tsoumani, IA Goudevenos, AD Tselepis
Expert Rev Clin Pharmacol. 2012;5(3):319-336.
http://www.medscape.com/viewarticle/764760
Abstract and Introduction
Cyclooxygenase-1 Inhibitors
P2Y12 Receptor Antagonists
GPIIb/IIIa Antagonists
Phosphodiesterase Inhibitors
Thrombin Receptor Antagonists
Thromboxane HYPERLINK “http://www.medscape.com/viewarticle/764760_7″&HYPERLINK “http://www.medscape.com/viewarticle/764760_7” Prostaglandin Endoperoxide Receptor Antagonists
Novel Antiplatelet Agents
Platelet activation plays a dominant role in hemostasis, a physiologic process that prevents blood loss when the vascular endothelium is injured. However, platelets also play a critical role in the formation of pathogenic thrombi in patients with atherothrombotic disease. Indeed, the disruption of an atherosclerotic plaque promotes platelet adhesion to various subendothelial matrix proteins such as von Willebrand factor (vWF) and collagen via glycoprotein (GP) receptors on the platelet surface (GPIbα–GPIX–GPV for vWF, GPVI–FcRγ and integrin α2β1 for collagen).
Platelet adhesion results in platelet activation and secretion of various soluble agonists, including ADP, thromboxane A2 (TxA2) and thrombin. These agonists bind to distinct G-protein-coupled receptors on the platelet surface, and induce further platelet activation, secretion and platelet aggregation, which is the result of the fibrinogen binding to the activated platelet integrin receptor αIIbβ3 (GPIIb/IIIa).
The currently available antiplatelet agents target key steps leading to platelet activation, including TxA2 synthesis, ADP-mediated signaling and GPIIb/IIIa-mediated platelet aggregation. Furthermore, ongoing research concerns the development of specific antagonists targeting receptors of other platelet agonists such as thrombin and TxA2. The management of patients who are receiving antiplatelet drugs requires a clear understanding of the pharmacokinetics and pharmacodynamics of these drugs, as well as their possible interaction with other drugs that may influence their therapeutic efficacy. In the present review, we provide an overview of the pharmacodynamic properties of all antiplatelet drugs used in daily clinical practice and discuss future perspectives on new antiplatelet agents under development.
Newer agents in antiplatelet therapy: a review
Abstract: Antiplatelet therapy remains the mainstay in preventing aberrant platelet activation in pathophysiological conditions such as myocardial infarction, ischemia, and stroke. Although there has been significant advancement in antiplatelet therapeutic approaches, aspirin still remains the gold standard treatment in the clinical setting. Limitations in safety, efficacy, and tolerability have precluded many of the antiplatelet inhibitors from use in patients. Unforeseen incidences of increased bleeding risk and recurrent arterial thrombosis observed in patients have hampered the development of superior next generation antiplatelet therapies. The pharmacokinetic and pharmacodynamic profiles have also limited the effectiveness of a number of antiplatelet inhibitors currently in use due to variability in metabolism, time to onset, and reversibility.
A focused effort in the development of newer antiplatelet therapies to address some of these shortcomings has resulted in a significant number of potential antiplatelet drugs which target enzymes (phosphodiesterase, cyclooxygenase), receptors (purinergic, prostaglandins, protease-activated receptors, thromboxane), and glycoproteins (αIIbβ3, GPVI, vWF, GPIb) in the platelet. The validation and search for newer antiplatelet therapeutic approaches proven to be superior to aspirin is still ongoing and should yield a better pharmacodynamic profile with fewer untoward side-effects to what is currently in use today.
Keywords: platelet aggregation inhibitors, blood platelets, purinergic P2Y receptor antagonists, receptor, PAR-1, platelet glycoprotein GPIIb-IIIa, thrombosis
Table 1. Approved antiplatelet drugs
Yeung and Holinstat
Leave a Reply