
Metabolic Genomics & Pharmaceutics
2015
http://www.amazon.com/dp/B012BB0ZF0
Author, Curator and Editor
Larry H Bernstein, MD, FCAP
Chief Scientific Officer
Leaders in Pharmaceutical Business Intelligence

Image Source: Courtesy of Google Images
Editor-in-Chief BioMed e-Series of e-Books
Leaders in Pharmaceutical Business Intelligence, Boston
avivalev-ari@alum.berkeley.edu
Other e-Books in the BioMedicine e-Series
Series A: e-Books on Cardiovascular Diseases
Content Consultant: Justin D Pearlman, MD, PhD, FACC
Volume One: Perspectives on Nitric Oxide
Sr. Editor: Larry Bernstein, MD, FCAP, Editor: Aviral Vatsa, PhD and Content Consultant: Stephen J Williams, PhD
available on Kindle Store @ Amazon.com
http://www.amazon.com/dp/B00DINFFYC
Volume Two: Cardiovascular Original Research: Cases in Methodology Design for Content Co-Curation
Curators: Justin D Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP, Aviva Lev-Ari, PhD, RN
- Causes
- Risks and Biomarkers
- Therapeutic Implications
Volume Three: Etiologies of CVD: Epigenetics, Genetics & Genomics
Curators: Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
- Causes
- Risks and Biomarkers
- Therapeutic Implications
Volume Four: Therapeutic Promise: CVD, Regenerative & Translational Medicine
Curators: Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
- Causes
- Risks and Biomarkers
- Therapeutic Implications
Volume Five: Pharmaco-Therapies for CVD
Curators: Justin D Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
- Causes
- Risks and Biomarkers
- Therapeutic Implications
Volume Six: Interventional Cardiology, Cardiac Surgery and Cardiovascular Imaging for Disease Diagnosis and Guidance of Treatment
Curators: Justin D Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
- Causes
- Risks and Biomarkers
- Therapeutic Implications
Series B: e-Books on Genomics & Medicine
Content Consultant: Larry H Bernstein, MD, FCAP
Volume 1: Genomics and Individualized Medicine
Sr. Editor: Stephen J Williams, PhD
Editors: Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Volume 2: Methodological Breakthroughs in NGS
Editor: Marcus Feldman, PhD, Prof. of Genetics, Stanford University
Volume 3: Institutional Leadership in Genomics
Editors: Marcus Feldman, PhD and Aviva Lev-Ari, PhD, RN
Series C: e-Books on Cancer & Oncology
Content Consultant: Larry H Bernstein, MD, FCAP
Volume 1: Cancer and Genomics
Sr. Editor: Stephen J Williams, PhD
Editors: Ritu Saxena, PhD, Tilda Barliya, PhD
Volume 2: Cancer Therapies: Metabolic, Genomics, Interventional, Immunotherapy and Nanotechnology in Therapy Delivery
Author, Curator and Editor: Larry H Bernstein, MD, FCAP
Guest Authors: Stephen J Williams, PhD, Dror Nir, PhD and Tilda Barliya, PhD, Demet Sag, PhD, Raphael Nir, PhD, Michael Briggs, PhD
Volume 3: Cancer Patients’ Resources on Therapies
Sr. Editor: TBA
Series D: e-Books on BioMedicine
Content Consultant: Larry H Bernstein, MD, FCAP
Volume 1: Metabolic Genomics & Pharmaceutics
Author, Curator and Editor: Larry H Bernstein, MD, FCAP
Volume 2: Infectious Diseases
Editor: TBA
Volume 3: Immunology and Therapeutics
Editor: TBA
Series E: Titles in the Strategic Plan for 2015
Volume 1: The Patient’s Voice: Personal Experience with Invasive Medical Procedures
Editor: TBA
Volume 2: Interviews with Scientific Leaders
Editor: TBA
Volume 3: Milestones in Physiology & Discoveries in Medicine and Genomics
Author, Curator and Editor: Larry H Bernstein, MD, FCAP
This e-Book is a comprehensive review of recent Original Research on METABOLOMICS and related opportunities for Targeted Therapy written by Experts, Authors, Writers. The results of Original Research are gaining value added for the e-Reader by the Methodology of Curation. The e-Book’s articles have been published on the Open Access Online Scientific Journal, since April 2012. All new articles on this subject, will continue to be incorporated, as published with periodical updates.
Open Access Online Journal
http://www.pharmaceuticalIntelligence.com
is a scientific, medical and business, multi-expert authoring environment for information syndication in several domains of Life Sciences, Medicine, Pharmaceutical and Healthcare Industries, BioMedicine, Medical Technologies & Devices. Scientific critical interpretations and original articles are written by PhDs, MDs, MD/PhDs, PharmDs, Technical MBAs as Experts, Authors, Writers (EAWs) on an Equity Sharing basis.
Metabolic Genomics & Pharmaceutics
Volume Author, Curator, Editor
Larry H Bernstein, MD, FCAP
electronic Table of Contents
Chapter 1: Metabolic Pathways
1.2 Studies of Respiration Lead to Acetyl CoA
1.3 Pentose Shunt, Electron Transfer, Galactose, more Lipids in brief
1.4 The Multi-step Transfer of Phosphate Bond and Hydrogen Exchange Energy
1.6 Glycosaminoglycans, Mucopolysaccharides, L-iduronidase, Enzyme Therapy
Chapter 2: Lipid Metabolism
2.1 Lipid Classification System
2.3 Lipid Oxidation and Synthesis of Fatty Acids
2.4 Cholesterol and Regulation of Liver Synthetic Pathways
2.5 Sex hormones, Adrenal cortisol, Prostaglandins
2.6 Cytoskeleton and Cell Membrane Physiology
2.7 Pharmacological Action of Steroid hormone
Chapter 3: Cell Signaling
3.1 Signaling and Signaling Pathways
3.2 Signaling Transduction Tutorial
3.3 Selected References to Signaling and Metabolic Pathways in Leaders in Pharmaceutical Intelligence
3.4 Integrins, Cadherins, Signaling and the Cytoskeleton
3.5 Complex Models of Signaling: Therapeutic Implications
3.6 Functional Correlates of Signaling Pathways
Chapter 4: Protein Synthesis and Degradation
4.1 The Role and Importance of Transcription Factors
4.2 RNA and the Transcription of the Genetic Code
4.4 Transcriptional Silencing and Longevity Protein Sir2
4.5 Ca2+ Signaling: Transcriptional Control
4.6 Long Noncoding RNA Network regulates PTEN Transcription
4.7 Zinc-Finger Nucleases (ZFNs) and Transcription Activator–Like Effector Nucleases (TALENs)
4.8 Cardiac Ca2+ Signaling: Transcriptional Control
4.9 Transcription Factor Lyl-1 Critical in Producing Early T-Cell Progenitors
4.10 Human Frontal Lobe Brain: Specific Transcriptional Networks
4.11 Somatic, Germ-cell, and Whole Sequence DNA in Cell Lineage and Disease
Chapter 5: Sub-cellular Structure
5.2 Mitochondrial Metabolism and Cardiac Function
5.3 Mitochondria: More than just the “Powerhouse of the Cell”
5.4 Mitochondrial Fission and Fusion: Potential Therapeutic Targets?
5.5 Mitochondrial Mutation Analysis might be “1-step” Away
5.7 Chromatophagy, A New Cancer Therapy: Starve The Diseased Cell Until It Eats Its Own DNA
5.9 Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility
Chapter 6: Proteomics
6.1 Proteomics, Metabolomics, Signaling Pathways, and Cell Regulation: a Compilation of Articles in the Journal http://pharmaceuticalintelligence.com
6.2 A Brief Curation of Proteomics, Metabolomics, and Metabolism
6.3 Using RNA-seq and Targeted Nucleases to Identify Mechanisms of Drug Resistance in Acute Myeloid Leukemia, SK Rathe in Nature, 2014
6.4 Proteomics – The Pathway to Understanding and Decision-making in Medicine
6.5 Advances in Separations Technology for the “OMICs” and Clarification of Therapeutic Targets
6.6 Expanding the Genetic Alphabet and Linking the Genome to the Metabolome
6.7 Genomics, Proteomics and Standards
6.8 Proteins and Cellular Adaptation to Stress
6.9 Genes, Proteomes, and their Interaction
6.10 Regulation of Somatic Stem Cell Function
6.11 Scientists discover that Pluripotency factor NANOG is also active in Adult Organism
Chapter 7: Metabolomics
7.1 Extracellular Evaluation of Intracellular Flux in Yeast Cells
7.2 Metabolomic Analysis of Two Leukemia Cell Lines Part I
7.3 Metabolomic Analysis of Two Leukemia Cell Lines Part II
7.6 Isoenzymes in Cell Metabolic Pathways
7.7 A Brief Curation of Proteomics, Metabolomics, and Metabolism
7.8 Metabolomics is about Metabolic Systems Integration
7.9 Mechanisms of Drug Resistance
7.10 Development Of Super-Resolved Fluorescence Microscopy
7.11 Metabolic Reactions Need Just Enough
Chapter 8. Impairments in Pathological States: Endocrine Disorders; Stress Hypermetabolism and CAncer
8.1 Omega3 Fatty Acids, Depleting the Source, and Protein Insufficiency in Renal Disease
8.2 Liver Endoplasmic Reticulum Stress and Hepatosteatosis
8.3 How Methionine Imbalance with Sulfur Insufficiency Leads to Hyperhomocysteinemia
8.4 AMPK Is a Negative Regulator of the Warburg Effect and Suppresses Tumor Growth InVivo
8.5 A Second Look at the Transthyretin Nutrition Inflammatory Conundrum
8.6 Mitochondrial Damage and Repair under Oxidative Stress
8.7 Metformin, Thyroid Pituitary Axis, Diabetes Mellitus, and Metabolism
8.8 Is the Warburg Effect the Cause or the Effect of Cancer: A 21st Century View?
8.9 Social Behavior Traits Embedded in Gene Expression
8.10 A Future for Plasma Metabolomics in Cardiovascular Disease Assessment
Chapter 9: Genomic Expression in Health and Disease
9.3 Metabolic Drivers in Aggressive Brain Tumors
9.4 Modified Yeast Produces a Range of Opiates for the First time
9.5 Parasitic Plant Strangleweed Injects Host With Over 9,000 RNA Transcripts
9.6 Plant-based Nutrition, Neutraceuticals and Alternative Medicine: Article Compilation the Journal
9.7 Reference Genes in the Human Gut Microbiome: The BGI Catalogue
9.8 Two Mutations, in the PCSK9 Gene: Eliminates a Protein involved in Controlling LDL Cholesterol
9.9 HDL-C: Target of Therapy – Steven E. Nissen, MD, MACC, Cleveland Clinic vs Peter Libby, MD, BWH
Summary
Epilogue
List of Contributors & Contributors’ Biographies
Chapter 1:
1.1 to 1.6
Chapter 2:
2.1 to 2.7
Chapter 3:
3.1 to 3.6
Chapter 4:
4.1 to 4.11
Chapter 5:
5.1 to 5.9
Chapter 6:
6.1 to 6.11
Chapter 7:
7.1 to 7.11
Chapter 8:
8.1 to 8.3. 8.5 – 8.10
Chapter 9:
9.6
Guest Authors, Curators and Reporters:
4.9, 9.3
5.3, 5.4, 5.5
5.9, 8.4
9.2
4.3, 4.10, 5.6, 5.7, 5.8, 5.9, 6.10, 7.5, 9.1, 9.4, 9.5, 9.7, 9.8, 9.9
Preface to Metabolomics as a Discipline in Medicine
Author: Larry H. Bernstein, MD, FCAP
Acknowledgements
I write this article in honor of my first mentor, Harry Maisel, Professor and Emeritus Chairman of Anatomy, Wayne State University, Detroit, MI and to my stimulating mentors, students, fellows, and associates over many years:
Masahiro Chiga, MD, PhD, Averill A Liebow, MD, Nathan O Kaplan, PhD, Johannes Everse, PhD, Norio Shioura, PhD, Abraham Braude, MD, Percy J Russell, PhD, Debby Peters, Walter D Foster, PhD, Herschel Sidransky, MD, Sherman Bloom, MD, Matthew Grisham, PhD, Christos Tsokos, PhD, IJ Good, PhD, Distinguished Professor, Raool Banagale, MD, Gustavo Reynoso, MD,Gustave Davis, MD, Marguerite M Pinto, MD, Walter Pleban, MD, Marion Feietelson-Winkler, RD, PhD, John Adan, MD, Joseph Babb, MD, Stuart Zarich, MD, Inder Mayall, MD, A Qamar, MD, Yves Ingenbleek, MD, PhD, Emeritus Professor, Bette Seamonds, PhD, Larry Kaplan, PhD, Pauline Y Lau, PhD, Gil David, PhD, Ronald Coifman, PhD, Emeritus Professor, Linda Brugler, RD, MBA, James Rucinski, MD, Gitta Pancer, Ester Engelman, Farhana Hoque, Mohammed Alam, Michael Zions, William Fleischman, MD, Salman Haq, MD, Jerard Kneifati-Hayek, Madeleine Schleffer, John F Heitner, MD, Arun Devakonda,MD, Liziamma George,MD, Suhail Raoof, MD, Charles Oribabor, MD, Anthony Tortolani, MD, Prof and Chairman, JRDS Rosalino, PhD, Aviva Lev Ari, PhD, RN, Rosser Rudolph, MD, PhD, Eugene Rypka, PhD, Jay Magidson, PhD, Izaak Mayzlin, PhD, Eli Kaplan, PhD, Richard Bing and Maurice Bernstein, PhD.
The family of ‘omics fields has rapidly outpaced its siblings over the decade since the completion of the Human Genome Project. It has derived much benefit from the development of Proteomics, which has recently completed a first draft of the human proteome. Since genomics, transcriptomics, and proteomics, have matured
considerably, it has become apparent that the search for a driver or drivers of cellular signaling and metabolic pathways could not depend on a full clarity of the genome. There have been unresolved issues, that are not solely comprehended from assumptions about mutations.
The most common diseases affecting mankind are derangements in metabolic pathways, develop at specific ages periods, and often in adulthood or in the geriatric period, and are at the intersection of signaling pathways. Moreover, the organs involved and systemic features are heavily influenced by physical activity, and by the air we breathe and the water we drink.
The emergence of the new science is also driven by a large body of work on protein structure, mechanisms of enzyme action, the modulation of gene expression, the pH dependent effects on protein binding and conformation.
Beyond what has just been said, a significant portion of DNA has been designated as “dark matter”. It turns out to have enormous importance in gene regulation, even though it is not transcriptional, effected in a modulatory way by “noncoding RNAs. Metabolomics is the comprehensive analysis of small molecule metabolites. These might be substrates of sequenced enzyme reactions, or they might be “inhibiting” RNAs just mentioned. In either case, they occur in the substructures of the cell called organelles, the cytoplasm, and in the cytoskeleton.
The reactions are orchestrated, and they can be modified with respect to the flow of metabolites based on pH, temperature, membrane structural modifications, and modulators. Since most metabolites are generated by
enzymatic proteins that result from gene expression, and metabolites give organisms their biochemical characteristics, the metabolome links genotype with phenotype.
Metabolomics is still developing, and the continued development has relied on two major events. The first is chromatographic separation and mass spectroscopy (MS), MS/MS, as well as advances in fluorescence
ultrasensitive optical photonic methods, and the second, as crucial, is the developments in computational biology. The continuation of this trend brings expectations of an impact on pharmaceutical and on neutraceutical developments, which will have an impact on medical practice. What has lagged behind, and may continue to contribute to the lag is the failure to develop a suitable electronic medical record to assist the physician in decisions confronted with so much as yet, hidden data, the ready availability of which could guide more effective
diagnosis and management of the patient. Put all of this together, and we can meet series challenges as the research community interprets and integrates the complex data they are acquiring.
Introduction to Metabolomics
Author: Larry H. Bernstein, MD, FCAP
This is the first volume of the Series D: e-Books on BioMedicine – Metabolomics, Immunology, Infectious Diseases. It is written for comprehension at the third year medical student level, or as a reference for licensing board exams, but it is also written for the education of a first time bachalaureate degree reader in the biological sciences. Hopefully, it can be read with great interest by the undergraduate student who is undecided in the choice of a career.
In the Preface, I failed to disclose that the term Metabolomics applies to plants, animals, bacteria, and both prokaryotes and eukaryotes. The metabolome for each organism is unique, but from an evolutionary perspective has metabolic pathways in common, and expressed in concert with the environment that these living creatures exist. The metabolome of each has adaptive accommodation with suppression and activation of pathways that are functional and necessary in balance, for its existence. Was it William Faulkner who said in his Nobel Prize acceptance that mankind shall not merely exist, but survive? That seems to be the overlying theme for all of life. If life cannot persist, a surviving “remnant” might continue. The history of life may well be etched into the genetic code, some of which is not expressed.
This work is apportioned into chapters in a sequence that is first directed at the major sources for the energy and the structure of life, in the carbohydrates, lipids, and fats, which are sourced from both plants and animals, and depending on their balance, results in an equilibrium, and a disequilibrium we refer to as disease. There is also a need to consider the nonorganic essentials which are derived from the soil, from water, and from the energy of the sun and the air we breathe, or in the case of water-bound metabolomes, dissolved gases.
In addition to the basic essential nutrients and their metabolic utilization, they are under cellular metabolic regulation that is tied to signaling pathways. In addition, the genetic expression of the organism is under regulatory control by the interaction of RNAs that interact with the chromatin genetic framework, with exosomes, and with protein modulators.This is referred to as epigenetics, but there are also drivers of metabolism that are shaped by the interactions between enzymes and substartes, and are related to the tertiary structure of a protein. The framework for diseases in a separate chapter. Pharmaceutical interventions that are designed to modulate specific metabolic targets are addressed as the pathways are unfolded. Neutraceuticals and plant based nutrition are covered in Chapter 8.
Chapter 1: Metabolic Pathways
Chapter 2. Lipid Metabolism
Chapter 3. Cell Signaling
Chapter 4. Protein Synthesis and Degradation
Chapter 5: Sub-cellular Structure
Chapter 6: Proteomics
Chapter 7: Metabolomics
Chapter 8. Impairments in Pathological States: Endocrine Disorders; Stress Hypermetabolism and Cancer
Chapter 1: Metabolic Pathways
Introduction to Metabolic Pathways
Author: Larry H. Bernstein, MD, FCAP
Humans, mammals, plants and animals, and eukaryotes and prokaryotes all share a common denominator in their manner of existence. It makes no difference whether they inhabit the land, or the sea, or another living host. They exist by virtue of their metabolic adaptation by way of taking in nutrients as fuel, and converting the nutrients to waste in the expenditure of carrying out the functions of motility, breakdown and utilization of fuel, and replication of their functional mass.
There are essentially two major sources of fuel, mainly, carbohydrate and fat. A third source, amino acids which requires protein breakdown, is utilized to a limited extent as needed from conversion of gluconeogenic amino acids for entry into the carbohydrate pathway. Amino acids follow specific metabolic pathways related to protein synthesis and cell renewal tied to genomic expression.
Carbohydrates are a major fuel utilized by way of either of two pathways. They are a source of readily available fuel that is accessible either from breakdown of disaccharides or from hepatic glycogenolysis by way of the Cori cycle. Fat derived energy is a high energy source that is metabolized by one carbon transfers using the oxidation of fatty acids in mitochondria. In the case of fats, the advantage of high energy is conferred by chain length.
Carbohydrate metabolism has either of two routes of utilization. This introduces an innovation by way of the mitochondrion or its equivalent, for the process of respiration, or aerobic metabolism through the tricarboxylic acid, or Krebs cycle. In the presence of low oxygen supply, carbohydrate is metabolized anaerobically, the six carbon glucose being split into two three carbon intermediates, which are finally converted from pyruvate to lactate. In the presence of oxygen, the lactate is channeled back into respiration, or mitochondrial oxidation, referred to as oxidative phosphorylation. The actual mechanism of this process was of considerable debate for some years until it was resolved that the mechanism involve hydrogen transfers along the “electron transport chain” on the inner membrane of the mitochondrion, and it was tied to the formation of ATP from ADP linked to the so called “active acetate” in Acetyl-Coenzyme A, discovered by Fritz Lipmann (and Nathan O. Kaplan) at Massachusetts General Hospital. Kaplan then joined with Sidney Colowick at the McCollum Pratt Institute at Johns Hopkins, where they shared tn the seminal discovery of the “pyridine nucleotide transhydrogenases” with Elizabeth Neufeld, who later established her reputation in the mucopolysaccharidoses (MPS) with L-iduronidase and lysosomal storage disease.
This chapter covers primarily the metabolic pathways for glucose, anaerobic and by mitochondrial oxidation, the electron transport chain, fatty acid oxidation, galactose assimilation, and the hexose monophosphate shunt, essential for the generation of NADPH. The is to be more elaboration on lipids and coverage of transcription, involving amino acids and RNA in other chapters.
The subchapters are as follows:
1.1 Carbohydrate Metabolism
1.2 Studies of Respiration Lead to Acetyl CoA
1.3 Pentose Shunt, Electron Transfer, Galactose, more Lipids in brief
1.4 The Multi-step Transfer of Phosphate Bond and Hydrogen Exchange Energy


1.1 Carbohydrate Metabolism
Larry H. Bernstein, MD, FCAP
1.2 Studies of Respiration Lead to Acetyl CoA
Larry H. Bernstein, MD, FCAP
1.3 Pentose Shunt, Electron Transfer, Galactose, more Lipids in brief
Larry H. Bernstein, MD, FCAP
1.4 The Multi-step Transfer of Phosphate Bond and Hydrogen Exchange Energy
Larry H. Bernstein, MD, FCAP
1.5 Diabetes Mellitus
Larry H. Bernstein, MD, FCAP
1.6 Glycosaminoglycans, Mucopolysaccharides, L-iduronidase, Enzyme Therapy
Larry H. Bernstein, MD, FCAP
Summary of Metabolic Pathways
Author and Curator: Larry H. Bernstein, MD, FCAP
This portion of a series of chapters on metabolism, proteomics and metabolomics dealt mainly with carbohydrate metabolism. Amino acids and lipids are presented more fully in the chapters that follow. There are features on the
- functioning of enzymes and proteins,
- on sequential changes in a chain reaction, and
- on conformational changes that we shall also cover.
These are critical to developing a more complete understanding of life processes.
I needed to lay out the scope of metabolic reactions and pathways, and their complementary changes. These may not appear to be adaptive, if the circumstances and the duration is not clear. The metabolic pathways map in total
is in interaction with environmental conditions – light, heat, external nutrients and minerals, and toxins – all of which give direction and strength to these reactions. A developing goal is to discover how views introduced by molecular biology and genomics don’t clarify functional cellular dynamics that are not related to the classical view. The work is vast.
Carbohydrate metabolism denotes the various biochemical processes responsible for the formation, breakdown and interconversion of carbohydrates in living organisms. The most important carbohydrate is glucose, a simple sugar (monosaccharide) that is metabolized by nearly all known organisms. Glucose and other carbohydrates are part of a wide variety of metabolic pathways across species: plants synthesize carbohydrates from carbon dioxide and water by photosynthesis storing the absorbed energy internally, often in the form of starch or lipids. Plant components are consumed by animals and fungi, and used as fuel for cellular respiration. Oxidation of one gram of carbohydrate yields approximately 4 kcal of energy and from lipids about 9 kcal. Energy obtained from metabolism (e.g. oxidation of glucose) is usually stored temporarily within cells in the form of ATP. Organisms capable of aerobic respiration metabolize glucose and oxygen to release energy with carbon dioxide and water as byproducts.
Carbohydrates are used for short-term fuel, and even though they are simpler to metabolize than fats, they don’t produce as equivalent energy yield measured by ATP. In animals, the concentration of glucose in the blood is linked to the pancreatic endocrine hormone, insulin. . In most organisms, excess carbohydrates are regularly catabolized to form acetyl-CoA, which is a feed stock for the fatty acid synthesis pathway; fatty acids, triglycerides, and other lipids are commonly used for long-term energy storage. The hydrophobic character of lipids makes them a much more compact form of energy storage than hydrophilic carbohydrates.
Glucose is metabolized obtaining ATP and pyruvate by way of first splitting a six-carbon into two three carbon chains, which are converted to lactic acid from pyruvate in the lactic dehydrogenase reaction. The reverse conversion is by a separate unidirectional reaction back to pyruvate after moving through pyruvate dehydrogenase complex.
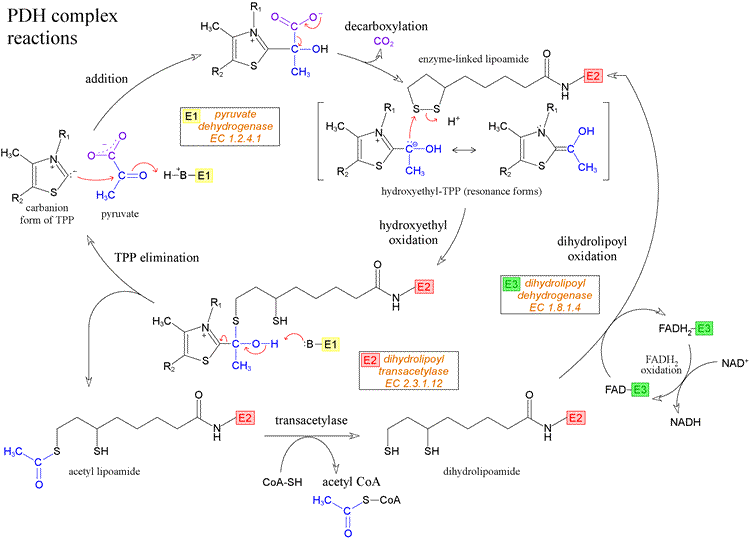
Pyruvate dehydrogenase complex (PDC) is a complex of three enzymes that convert pyruvate into acetyl-CoA by a process called pyruvate decarboxylation. Acetyl-CoA may then be used in the citric acid cycle to carry out cellular respiration, and this complex links the glycolysis metabolic pathway to the citric acid cycle. This multi-enzyme complex is related structurally and functionally to the oxoglutarate dehydrogenase and branched-chain oxo-acid dehydrogenase multi-enzyme complexes. In eukaryotic cells the reaction occurs inside the mitochondria, after transport of the substrate, pyruvate, from the cytosol. The transport of pyruvate into the mitochondria is via a transport protein and is active, consuming energy. On entry to the mitochondria pyruvate decarboxylation occurs, producing acetyl CoA. This irreversible reaction traps the acetyl CoA within the mitochondria. Pyruvate dehydrogenase deficiency from mutations in any of the enzymes or cofactors results in lactic acidosis.

http://guweb2.gonzaga.edu/faculty/cronk/biochem/images/PDH-rxns.gif
{kind=link}
Typically, a breakdown of one molecule of glucose by aerobic respiration (i.e. involving both glycolysis and Kreb’s cycle) is about 33-35 ATP. This is categorized as:
Glycogenolysis – the breakdown of glycogen into glucose, which provides a glucose supply for glucose-dependent tissues.
Glycogenolysis in liver provides circulating glucose short term.
Glycogenolysis in muscle is obligatory for muscle contraction.
Pyruvate from glycolysis enters the Krebs cycle, also known as the citric acid cycle, in aerobic organisms.
Anaerobic breakdown by glycolysis – yielding 8-10 ATP
Aerobic respiration by Kreb’s cycle – yielding 25 ATP
The pentose phosphate pathway (shunt) converts hexoses into pentoses and regenerates NADPH. NADPH is an essential antioxidant in cells which prevents oxidative damage and acts as precursor for production of many biomolecules.
Glycogenesis – the conversion of excess glucose into glycogen as a cellular storage mechanism; achieving low osmotic pressure.
Gluconeogenesis – de novo synthesis of glucose molecules from simple organic compounds. An example in humans is the conversion of a few amino acids in cellular protein to glucose.
Metabolic use of glucose is highly important as an energy source for muscle cells and in the brain, and red blood cells.
The hormone insulin is the primary glucose regulatory signal in animals. It mainly promotes glucose uptake by the cells, and it causes the liver to store excess glucose as glycogen. Its absence
- turns off glucose uptake,
- reverses electrolyte adjustments,
- begins glycogen breakdown and glucose release into the circulation by some cells,
- begins lipid release from lipid storage cells, etc.
The level of circulatory glucose (known informally as “blood sugar”) is the most important signal to the insulin-producing cells.
- insulin is made by beta cells in the pancreas,
- fat is stored n adipose tissue cells, and
- glycogen is both stored and released as needed by liver cells.
- no glucose is released to the blood from internal glycogen stores from muscle cells.
The hormone glucagon, on the other hand, opposes that of insulin, forcing the conversion of glycogen in liver cells to glucose, and then release into the blood. Growth hormone, cortisol, and certain catecholamines (such as epinepherine) have glucoregulatory actions similar to glucagon. These hormones are referred to as stress hormones because they are released under the influence of catabolic proinflammatory (stress) cytokines – interleukin-1 (IL1) and tumor necrosis factor α (TNFα).
| Net Yield of GlycolysisThe preparatory phase consumes 2 ATPThe pay-off phase produces 4 ATP.The gross yield of glycolysis is therefore4 ATP – 2 ATP = 2 ATPThe pay-off phase also produces 2 molecules of NADH + H+ which can be further converted to a total of 5 molecules of ATP* by the electron transport chain (ETC) during oxidative phosphorylation.Thus the net yield during glycolysis is 7 molecules of ATP* This is calculated assuming one NADH molecule gives 2.5 molecules of ATP during oxidative phosphorylation. |
Cellular respiration involves 3 stages for the breakdown of glucose – glycolysis, Kreb’s cycle and the electron transport system. Kreb’s cycle produces about 60-70% of ATP for release of energy in the body. It directly or indirectly connects with all the other individual pathways in the body.
| The Kreb’s Cycle occurs in two stages: |
- Conversion of Pyruvate to Acetyl CoA
- Acetyl CoA Enters the Kreb’s Cycle
Each pyruvate in the presence of pyruvate dehydrogenase (PDH) complex in the mitochondria gets converted to acetyl CoA which in turn enters the Kreb’s cycle. This reaction is called as oxidative decarboxylation as the carboxyl group is removed from the pyruvate molecule in the form of CO2 thus yielding 2-carbon acetyl group which along with the coenzyme A forms acetyl CoA.
The PDH requires the sequential action of five co-factors or co-enzymes for the combined action of dehydrogenation and decarboxylation to take place. These five are TPP (thiamine phosphate), FAD (flavin adenine dinucleotide), NAD (nicotinamide adenine dinucleotide), coenzyme A (denoted as CoA-SH at times to depict role of -SH group) and lipoamide.
Acetyl CoA condenses with oxaloacetate (4C) to form a citrate (6C) by transferring its acetyl group in the presence of enzyme citrate synthase. The CoA liberated in this reaction is ready to participate in the oxidative decarboxylation of another molecule of pyruvate by PDH complex.
Isocitrate undergoes oxidative decarboxylation by the enzyme isocitrate dehydrogenase to form oxalosuccinate (intermediate- not shown) which in turn forms α-ketoglutarate (also known as oxoglutarate) which is a five carbon compound. CO2 and NADH are released in this step. α-ketoglutarate (5C) undergoes oxidative decarboxylation once again to form succinyl CoA (4C) catalysed by the enzyme α-ketoglutarate dehydrogenase complex.
Succinyl CoA is then converted to succinate by succinate thiokinase or succinyl coA synthetase in a reversible manner. This reaction involves an intermediate step in which the enzyme gets phosphorylated and then the phosphoryl group which has a high group transfer potential is transferred to GDP to form GTP.
Succinate then gets oxidised reversibly to fumarate by succinate dehydrogenase. The enzyme contains iron-sulfur clusters and covalently bound FAD which when undergoes electron exchange in the mitochondria causes the production of FADH2.
Fumarate is then by the enzyme fumarase converted to malate by hydration (addition of H2O) in a reversible manner.
Malate is then reversibly converted to oxaloacetate by malate dehydrogenase which is NAD linked and thus produces NADH.
The oxaloacetate produced is now ready to be utilized in the next cycle by the citrate synthase reaction and thus the equilibrium of the cycle shifts to the right.
The NADH formed in the cytosol can yield variable amounts of ATP depending on the shuttle system utilized to transport them into the mitochondrial matrix. This NADH, formed in the cytosol, is impermeable to the mitochondrial inner-membrane where oxidative phosphorylation takes place. Thus to carry this NADH to the mitochondrial matrix there are special shuttle systems in the body. The most active shuttle is the malate-aspartate shuttle via which 2.5 molecules of ATP are generated for 1 NADH molecule. This shuttle is mainly used by the heart, liver and kidneys. The brain and skeletal muscles use the other shuttle known as glycerol 3-phosphate shuttle which synthesizes 1.5 molecules of ATP for 1 NADH.
Glucose-6-phosphate Dehydrogenase is the committed step of the Pentose Phosphate Pathway. This enzyme is regulated by availability of the substrate NADP+. As NADPH is utilized in reductive synthetic pathways, the increasing concentration of NADP+ stimulates the Pentose Phosphate Pathway, to replenish NADPH. The importance of this pathway can easily be underestimated. The main source for energy in respiration was considered to be tied to the high energy phosphate bond in phosphorylation and utilizes NADPH, converting it to NADP+. The pentose phosphate shunt is essential for the generation of nucleic acids, in regeneration of red cells and lens – requiring NADPH.
NAD+ serves as electron acceptor in catabolic pathways in which metabolites are oxidized. The resultant NADH is reoxidized by the respiratory chain, producing ATP.
The pyridine nucleotide transhydrogenase reaction concerns the energy-dependent reduction of TPN by DPNH. In 1959, Klingenberg and Slenczka made the important observation that incubation of isolated liver mitochondria with DPN-specific substrates or succinate in the absence of phosphate acceptor resulted in a rapid and almost complete reduction of the intramitochondrial TPN. These and related findings led Klingenberg and co-workers (1-3) to postulate the occurrence of a ATP-controlled transhydrogenase reaction catalyzing the reduction of TPN by DPNH. (The role of transhydrogenase in the energy-linked reduction of TPN. Fritz Hommes, Ronald W. Estabrook, The Wenner-Gren Institute, University of Stockholm, Stockholm, Sweden. Biochemical and Biophysical Research Communications 11, (1), 2 Apr 1963, Pp 1–6.
http://dx.doi.org:/10.1016/0006-291X(63)90017-2/).
Further studies observed the coupling of TPN-specific dehydrogenases with the transhydrogenase and observing the reduction of large amounts of diphosphopyridine nucleotide (DPN) in the presence of catalytic amounts of triphosphopyridine nucleotide (TPN). The studies showed the direct interaction between TPNHz and DPN, in the presence of transhydrogenase to yield products having the properties of TPN and DPNHZ. The reaction involves a transfer of electrons (or hydrogen) rather than a phosphate. (Pyridine Nucleotide Transhydrogenase II. Direct Evidence for and Mechanism of the Transhydrogenase Reaction* by Nathan 0. Kaplan, Sidney P. Colowick, And Elizabeth F. Neufeld. (From The Mccollum-Pratt Institute, The Johns Hopkins University, Baltimore, Maryland) J. Biol. Chem. 1952, 195:107-119.)
http://www.JBC.org/Content/195/1/107.Citation
Notation: TPN, NADP; DPN, NAD+; reduced pyridine nucleotides: TPNH (NADPH2), DPNH (NADH).
Note: In this discussion there is a detailed presentation of the activity of lactic acid conversion in the mitochondria by way of PDH. In a later section there is mention of the bidirectional reaction of lactate dehydrogenase. However, the forward reaction is dominant (pyruvate to lactate) and is described. This is not related to the kinetics of the LD reaction with respect to the defining characteristic – Km.
Biochemical Education Jan 1977; 5(1):15. Kinetics of Lactate Dehydrogenase: A Textbook Problem.
K.L. MANCHESTER. Department of Biochemistry, University of Witwatersrand, Johannesburg South Africa.
One presupposes that determined Km values are meaningful under intracellular conditions. In relation to teaching it is a simple experiment for students to determine for themselves the Km towards pyruvate of LDH in a post-mitochondrial supernatant of rat heart and thigh muscle. The difference in Km may be a factor of 3 or 4-fold.It is pertinent then to ask what is the range of suhstrate concentrations over which a difference in Km may be expected to lead to significant differences in activity and how these concentrations compare with pyruvate concentrations in the cell. The evidence of Vesell and co-workers that inhibition by pyruvate is more readily seen at low than at high enzyme concentration is important in emphasizing that under intracellular conditions enzyme concentrations may be relatively large in relation to the substrate available. This will be particularly so in relation to [NADH] which in the cytoplasm is likely to be in the ~M range.
A final point concerns the kinetic parameters for LDH quoted by Bergmeyer for lactate estimations a pH of 9 is recommended and the Km towards lactate at that pH is likely to be appreciably different from the quoted values at pH 7 — Though still at pH 9 showing a substantially lower value for lactate with the heart preparation. http://onlinelibrary.wiley.com/doi/10.1016/0307-4412%2877%2990013-9/pdf
Several investigators have established that epidermis converts most of the glucose it uses to lactic acid even in the presence of oxygen. This is in contrast to most tissues where lactic acid production is used for energy production only when oxygen is not available. This large amount of lactic acid being continually produced within the epidermal cell must be excreted by the cell and then carried away by the blood stream to other tissues where the lactate can be utilized. The LDH reaction with pyruvate and NADH is reversible although at physiological pH the equilibrium position for the reaction lies very far to the right, i.e., in favor of lactate production. The speed of this reaction depends not only on the amount of enzyme present but also on the concentrations of the substances involved on both sides of the equation. The net direction in which the reaction will proceed depends solely on the relative concentrations of the substances on each side of the equation.
In vivo there is net conversion of pyruvate (formed from glucose) to lactate. Measurements of the speed of lactate production by sheets of epidermis floating on a medium containing glucose indicate a rate of lactate production of approximately 0.7 rn/sm/mm/mg of fresh epidermis.Slice incubation experiments are presumably much closer to the actual in vivo conditions than the homogenate experiments. The discrepancy between the two indicates that in vivo conditions are far from optimal for the conversion of pyruvate to lactate. Only 1/100th of the maximal activity of the enzyme present is being achieved. The concentrations of the various substances involved are not
optimal in vivo since pyruvate and NADH concentrations are lower than lactate and NAD concentrations and this might explain the in vivoinhibition of LDH activity. (Lactate Production And Lactate Dehydrogenase In The Human Epidermis*. KM. Halprin, A Ohkawara. J Invest Dermat 1966; 47(3): 222-6.)
http://www.nature.com/jid/journal/v47/n3/pdf/jid1966133a.pdf
Chapter 2. Lipid Metabolism
Introduction to Lipid Metabolism
Author: Larry H. Bernstein, MD, FCAP
This series of articles is concerned with lipid metabolism. These discussions lay the groundwork to proceed to discussions that will take on a somewhat different approach, but they are critical to developing a more complete point of view of life processes. I have indicated that there are protein-protein interactions or protein-membrane interactions and associated regulatory features, but the focus of the discussion or points made were different, and will be returned to. The role of lipids in circulating plasma proteins as biomarkers for coronary vascular disease
can be traced to the early work of Frederickson and the classification of lipid disorders. The very critical role of lipids in membrane structure in health and disease has had much less attention, despite the enormous importance,
especially in the nervous system.
This portion of the discussions of metabolism will have several topics on lipid metabolism. The first is concerned with the basic types of lipids -which are defined structurally and have different carbon chain length, and have
two basic types of indispensible fatty acid derivations – along pro-inflammatory
and anti-inflammatory pathways:
- Alpha-linolenic acid (ALA) and linoleic acid (LA), n-3 polyunsaturated fatty acids LCPUFAs (EPA, DHA, and AA), eicosanoids,
delta-3-desaturase, prostaglandins, and leukotrienes. - the role of the mitochondrial electron transport chain in hydrogen transfers
and oxidative phosphorylation with respect to the oxidation of fatty acids
and fatty acid synthesis. - The membrane structures of the cell, including
- the cytoskeleton, essential organelles, and the intercellular matrix, which
is a critical consideration for - cell motility, membrane conductivity, flexibility, and signaling.
- The membrane structure involves aggregation of lipids with proteins,
- and is associated with hydrophobicity.
- The pathophysiology of systemic circulating lipid disorders.
- The fifth is the pathophysiology of cell structures under oxidative
stress. - Lipid disposal and storage diseases.
Author: Larry H. Bernstein, MD, FCAP
Lipid Classification System
The LIPID MAPS Lipid Classification System is comprised of eight lipid categories, each with its own sublassification hierarchy.
http://www.lipidmaps.org/resources/tutorials/lipid_cns.html
Each LMSD record contains an image of the
- molecular structure,
- common and systematic names,
- links to external databases,
- Wikipedia pages (where available),
- other annotations and links to structure viewing tools.
All lipids in the LIPID MAPS Structure Database (LMSD) have been classified using this system and have been assigned LIPID MAPS ID’s (LM_ID) which reflects their position in the classification hierarchy.
The LIPID MAPS Structure Database (LMSD) is a relational database encompassing structures and annotations of biologically relevant lipids. As of May 3, 2013, LMSD contains over 37,500 unique lipid structures, making it the largest public lipid-only database in the world. Structures of lipids in the database come from several sources:
- LIPID MAPS Consortium’s core laboratories and partners;
- lipids identified by LIPID MAPS experiments;
- biologically relevant lipids manually curated from LIPID BANK, LIPIDAT, Lipid Library, Cyberlipids, ChEBI and other public sources;
- novel lipids submitted to peer-reviewed journals;
- computationally generated structures for appropriate classes.
All the lipid structures in LMSD adhere to the structure drawing rules proposed by the LIPID MAPS consortium. A number of structure viewing options are offered: gif image (default), Chemdraw (requires Chemdraw ActiveX/Plugin), MarvinView (Java applet) and JMol (Java applet).
(as of 10/8/14)
Number of lipids per category
Fatty acyls 5869
Glycerolipids 7541
Glycerophospholipids 8002
Sphingolipids 4338
Sterol lipids 2715
Prenol lipids 1259
Sacccharolipids 1293
Polyketides 6742
TOTAL 37,759 structures
References
Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, Merrill AH Jr, Murphy RC, Raetz CR, Russell DW, Subramaniam S. LMSD: LIPID MAPS structure database Nucleic Acids Research 35: p. D527-32. PMID:17098933 [doi:10.1093/nar/gkl838] PMID: 17098933
Fahy E, Sud M, Cotter D & Subramaniam S. LIPID MAPS online tools for lipid research Nucleic Acids Research (2007) 35: p. W606-12.PMID:17584797 [doi:10.1093/nar/gkm324] PMID: 17584797
The Recognition of Essential Fatty Acids
Dietary fat has long been recognized as an important source of energy for mammals, but in the late 1920s, researchers demonstrated the dietary requirement for particular fatty acids, which came to be called essential fatty acids. It was not until the advent of intravenous feeding, however, that the importance of essential fatty acids was widely accepted: Clinical signs of essential fatty acid deficiency are generally observed only in patients on total parenteral nutrition who received mixtures devoid of essential fatty acids or in those with malabsorption syndromes.
These signs include dermatitis and changes in visual and neural function. Over the past 40 years, an increasing number of physiological functions, such as immunomodulation, have been attributed to the essential fatty acids and their metabolites, and this area of research remains quite active.1, 2
Fatty Acid Nomenclature
The fat found in foods consists largely of a heterogeneous mixture of triacylglycerols (triglycerides)–glycerol molecules that are each combined with three fatty acids. The fatty acids can be divided into two categories, based on chemical properties: saturated fatty acids, which are usually solid at room temperature, and unsaturated fatty acids, which are liquid at room temperature. The term “saturation” refers to a chemical structure in which each carbon atom in the fatty acyl chain is bound to (saturated with) four other atoms, these carbons are linked by single bonds, and no other atoms or molecules can attach; unsaturated fatty acids contain at least one pair of carbon atoms linked by a double bond, which allows the attachment of additional atoms to those carbons (resulting in saturation). Despite their differences in structure, all fats contain approximately the same amount of energy (37 kilojoules/gram, or 9 kilocalories/gram).
The class of unsaturated fatty acids can be further divided into monounsaturated and polyunsaturated fatty acids. Monounsaturated fatty acids (the primary constituents of olive and canola oils) contain only one double bond. Polyunsaturated fatty acids (PUFAs) (the primary constituents of corn, sunflower, flax seed and many other vegetable oils) contain more than one double bond. Fatty acids are often referred to using the number of carbon atoms in the acyl chain, followed by a colon, followed by the number of double bonds in the chain (e.g., 18:1 refers to the 18-carbon monounsaturated fatty acid, oleic acid; 18:3 refers to any 18-carbon PUFA with three double bonds).
PUFAs are further categorized on the basis of the location of their double bonds. An omega or n notation indicates the number of carbon atoms from the methyl end of the acyl chain to the first double bond. Thus, for example, in the omega-3 (n-3) family of PUFAs, the first double bond is 3 carbons from the methyl end of the molecule. Finally, PUFAs can be categorized according to their chain length. The 18-carbon n-3 and n-6 short-chain PUFAs are precursors to the longer 20- and 22-carbon PUFAs, called long-chain PUFAs (LCPUFAs).
Fatty Acid Metabolism
Mammalian cells can introduce double bonds into all positions on the fatty acid chain except the n-3 and n-6 position. Thus, the short-chain alpha- linolenic acid (ALA, chemical abbreviation: 18:3n-3) and linoleic acid (LA, chemical abbreviation: 18:2n-6) are essential fatty acids.
No other fatty acids found in food are considered ‘essential’ for humans, because they can all be synthesized from the short chain fatty acids.
Following ingestion, ALA and LA can be converted in the liver to the long chain, more unsaturated n-3 and n-6 LCPUFAs by a complex set of synthetic pathways that share several enzymes (Figure 1). LC PUFAs retain the original sites of desaturation (including n-3 or n-6). The omega-6 fatty acid LA is converted to gamma-linolenic acid (GLA, 18:3n-6), an omega- 6 fatty acid that is a positional isomer of ALA. GLA, in turn, can be converted to the longerchain omega-6 fatty acid, arachidonic acid (AA, 20:4n-6). AA is the precursor for certain classes of an important family of hormone- like substances called the eicosanoids (see below).
The omega-3 fatty acid ALA (18:3n-3) can be converted to the long-chain omega-3 fatty acid, eicosapentaenoic acid (EPA; 20:5n-3). EPA can be elongated to docosapentaenoic acid (DPA 22:5n-3), which is further desaturated to docosahexaenoic acid (DHA; 22:6n-3). EPA and DHA are also precursors of several classes of eicosanoids and are known to play several other critical roles, some of which are discussed further below.
The conversion from parent fatty acids into the LC PUFAs – EPA, DHA, and AA – appears to occur slowly in humans. In addition, the regulation of conversion is not well understood, although it is known that ALA and LA compete for entry into the metabolic pathways.
Physiological Functions of EPA and AA
As stated earlier, fatty acids play a variety of physiological roles. The specific biological functions of a fatty acid are determined by the number and position of double bonds and the length of the acyl chain.
Both EPA (20:5n-3) and AA (20:4n-6) are precursors for the formation of a family of hormone- like agents called eicosanoids. Eicosanoids are rudimentary hormones or regulating – molecules that appear to occur in most forms of life. However, unlike endocrine hormones, which travel in the blood stream to exert their effects at distant sites, the eicosanoids are autocrine or paracrine factors, which exert their effects locally – in the cells that synthesize them or adjacent cells. Processes affected include the movement of calcium and other substances into and out of cells, relaxation and contraction of muscles, inhibition and promotion of clotting, regulation of secretions including digestive juices and hormones, and control of fertility, cell division, and growth.3
The eicosanoid family includes subgroups of substances known as prostaglandins, leukotrienes, and thromboxanes, among others. As shown in Figure 1.1, the long-chain omega-6 fatty acid, AA (20:4n-6), is the precursor of a group of eicosanoids that include series-2 prostaglandins and series-4 leukotrienes. The omega-3 fatty acid, EPA (20:5n-3), is the precursor to a group of eicosanoids that includes series-3 prostaglandins and series-5 leukotrienes. The AA-derived series-2 prostaglandins and series-4 leukotrienes are often synthesized in response to some emergency such as injury or stress, whereas the EPA-derived series-3 prostaglandins and series-5 leukotrienes appear to modulate the effects of the series-2 prostaglandins and series-4 leukotrienes (usually on the same target cells). More specifically, the series-3 prostaglandins are formed at a slower rate and work to attenuate the effects of excessive levels of series-2 prostaglandins. Thus, adequate production of the series-3 prostaglandins seems to protect against heart attack and stroke as well as certain inflammatory diseases like arthritis, lupus, and asthma.3.
EPA (22:6 n-3) also affects lipoprotein metabolism and decreases the production of substances – including cytokines, interleukin 1ß (IL-1ß), and tumor necrosis factor a (TNF-a) – that have pro-inflammatory effects (such as stimulation of collagenase synthesis and the expression of adhesion molecules necessary for leukocyte extravasation [movement from the circulatory system into tissues]).2 The mechanism responsible for the suppression of cytokine production by omega-3 LC PUFAs remains unknown, although suppression of omega-6-derived eicosanoid production by omega-3 fatty acids may be involved, because the omega-3 and omega-6 fatty acids compete for a common enzyme in the eicosanoid synthetic pathway, delta-6 desaturase.
DPA (22:5n-3) (the elongation product of EPA) and its metabolite DHA (22:6n-3) are frequently referred to as very long chain n-3 fatty acids (VLCFA). Along with AA, DHA is the major PUFA found in the brain and is thought to be important for brain development and function. Recent research has focused on this role and the effect of supplementing infant formula with DHA (since DHA is naturally present in breast milk but not in formula).
Overview of Lipid Catabolism:
http://www.elmhurst.edu/~chm/vchembook/622overview.html
The major aspects of lipid metabolism are involved with
- Fatty Acid Oxidation to produce energy or
- the synthesis of lipids which is called Lipogenesis.
The metabolism of lipids and carbohydrates are related by the conversion of lipids from carbohydrates. This can be seen in the diagram. Notice the link through actyl-CoA, the seminal discovery of Fritz Lipmann. The metabolism of both is upset by diabetes mellitus, which results in the release of ketones (2/3 betahydroxybutyric acid) into the circulation.

http://www.elmhurst.edu/~chm/vchembook/images/590metabolism.gif
{kind=link}
The first step in lipid metabolism is the hydrolysis of the lipid in the cytoplasm to produce glycerol and fatty acids.
Since glycerol is a three carbon alcohol, it is metabolized quite readily into an intermediate in glycolysis, dihydroxyacetone phosphate. The last reaction is readily reversible if glycerol is needed for the synthesis of a lipid.
The hydroxyacetone, obtained from glycerol is metabolized into one of two possible compounds. Dihydroxyacetone may be converted into pyruvic acid, a 3-C intermediate at the last step of glycolysis to make energy.
In addition, the dihydroxyacetone may also be used in gluconeogenesis (usually dependent on conversion of gluconeogenic amino acids) to make glucose-6-phosphate for glucose to the blood or glycogen depending upon what is required at that time.
Fatty acids are oxidized to acetyl CoA in the mitochondria using the fatty acid spiral. The acetyl CoA is then ultimately converted into ATP, CO2, and H2O using the citric acid cycle and the electron transport chain.
There are two major types of fatty acids – ω-3 and ω-6. There are also saturated and unsaturated with respect to the existence of double bonds, and monounsaturated and polyunsatured. Polyunsaturated fatty acids (PUFAs) are important in long term health, and it will be seen that high cardiovascular risk is most associated with a low ratio of ω-3/ω-6, the denominator being from animal fat. Ω-3 fatty acids are readily available from fish, seaweed, and flax seed. More can be said of this later.
Fatty acids are synthesized from carbohydrates and occasionally from proteins. Actually, the carbohydrates and proteins have first been catabolized into acetyl CoA. Depending upon the energy requirements, the acetyl CoA enters the citric acid cycle or is used to synthesize fatty acids in a process known as LIPOGENESIS.
The relationships between lipid and carbohydrate metabolism are
summarized in Figure 2.

Energy Production Fatty Acid Oxidation:
“Visible” ATP:
In the fatty acid spiral, there is only one reaction which directly uses ATP and that is in the initiating step. So this is a loss of ATP and must be subtracted later.
A large amount of energy is released and restored as ATP during the oxidation of fatty acids. The ATP is formed from both the fatty acid spiral and the citric acid cycle.
Connections to Electron Transport and ATP:
One turn of the fatty acid spiral produces ATP from the interaction of the coenzymes FAD (step 1) and NAD+ (step 3) with the electron transport chain. Total ATP per turn of the fatty acid spiral is:
Electron Transport Diagram – (e.t.c.)
Step 1 – FAD into e.t.c. = 2 ATP
Step 3 – NAD+ into e.t.c. = 3 ATP
Total ATP per turn of spiral = 5 ATP
In order to calculate total ATP from the fatty acid spiral, you must calculate the number of turns that the spiral makes. Remember that the number of turns is found by subtracting one from the number of acetyl CoA produced. See the graphic on the left bottom.
Example with Palmitic Acid = 16 carbons = 8 acetyl groups
Number of turns of fatty acid spiral = 8-1 = 7 turns
ATP from fatty acid spiral = 7 turns and 5 per turn = 35 ATP.
This would be a good time to remember that single ATP that was needed to get the fatty acid spiral started. Therefore subtract it now.
NET ATP from Fatty Acid Spiral = 35 – 1 = 34 ATP
SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver
Jay D. Horton1,2, Joseph L. Goldstein1 and Michael S. Brown1
1Department of Molecular Genetics, and
2Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA
J Clin Invest. 2002;109(9):1125–1131.
http://dx.doi.org:/10.1172/JCI15593
Lipid homeostasis in vertebrate cells is regulated by a family of membrane-bound transcription factors designated sterol regulatory element–binding proteins (SREBPs). SREBPs directly activate the expression of more than 30 genes dedicated to the synthesis and uptake of cholesterol, fatty acids, triglycerides, and phospholipids, as well as the NADPH cofactor required to synthesize these molecules (1–4). In the liver, three SREBPs regulate the production of lipids for export into the plasma as lipoproteins and into the bile as micelles. The complex, interdigitated roles of these three SREBPs have been dissected through the study of ten different lines of gene-manipulated mice. These studies form the subject of this review.
SREBPs: activation through proteolytic processing
SREBPs belong to the basic helix-loop-helix–leucine zipper (bHLH-Zip) family of transcription factors, but they differ from other bHLH-Zip proteins in that they are synthesized as inactive precursors bound to the endoplasmic reticulum (ER) (1, 5). Each SREBP precursor of about 1150 amino acids is organized into three domains: (a) an NH2-terminal domain of about 480 amino acids that contains the bHLH-Zip region for binding DNA; (b) two hydrophobic transmembrane–spanning segments interrupted by a short loop of about 30 amino acids that projects into the lumen of the ER; and (c) a COOH-terminal domain of about 590 amino acids that performs the essential regulatory function described below.
In order to reach the nucleus and act as a transcription factor, the NH2-terminal domain of each SREBP must be released from the membrane proteolytically (Figure1). Three proteins required for SREBP processing have been delineated in cultured cells, using the tools of somatic cell genetics (see ref. 5for review). One is an escort protein designated SREBP cleavage–activating protein (SCAP). The other two are proteases, designated Site-1 protease (S1P) and Site-2 protease (S2P). Newly synthesized SREBP is inserted into the membranes of the ER, where its COOH-terminal regulatory domain binds to the COOH-terminal domain of SCAP (Figure 1).
Figure 1

Model for the sterol-mediated proteolytic release of SREBPs from membranes. SCAP is a sensor of sterols and an escort of SREBPs. When cells are depleted of sterols, SCAP transports SREBPs from the ER to the Golgi apparatus, where two proteases, Site-1 protease (S1P) and Site-2 protease (S2P), act sequentially to release the NH2-terminal bHLH-Zip domain from the membrane. The bHLH-Zip domain enters the nucleus and binds to a sterol response element (SRE) in the enhancer/promoter region of target genes, activating their transcription.
SCAP is both an escort for SREBPs and a sensor of sterols. When cells become depleted in cholesterol, SCAP escorts the SREBP from the ER to the Golgi apparatus, where the two proteases reside. In the Golgi apparatus, S1P, a membrane-bound serine protease, cleaves the SREBP in the luminal loop between its two membrane-spanning segments, dividing the SREBP molecule in half. (Fig 1) The NH2-terminal bHLH-Zip domain is then released from the membrane via a second cleavage mediated by S2P, a membrane-bound zinc metalloproteinase. The NH2-terminal domain, designated nuclear SREBP (nSREBP), translocates to the nucleus, where it activates transcription by binding to nonpalindromic sterol response elements (SREs) in the promoter/enhancer regions of multiple target genes.
SREBPs: two genes, three proteins
The mammalian genome encodes three SREBP isoforms, designated SREBP-1a, SREBP-1c, and SREBP-2.
SREBP-1a is a potent activator of all SREBP-responsive genes, including those that mediate the synthesis of cholesterol, fatty acids, and triglycerides. High-level transcriptional activation is dependent on exon 1a, which encodes a longer acidic transactivation segment than does the first exon of SREBP-1c. The roles of SREBP-1c and SREBP-2 are more restricted than that of SREBP-1a. SREBP-1c preferentially enhances transcription of genes required for fatty acid synthesis but not cholesterol synthesis.
SREBP-1c and SREBP-2 activate three genes required to generate NADPH, which is consumed at multiple stages in these lipid biosynthetic pathways (8) (Figure 2).

Steroids
A major class of lipids, steroids, have a ring structure of three cyclohexanes and one
cyclopentane in a fused ring system as shown below. There are a variety of functional
groups that may be attached. The main feature, as in all lipids, is the large number of
carbon-hydrogens which make steroids non-polar.
Steroids include such well known compounds as cholesterol, sex hormones, birth
control pills, cortisone, and anabolic steroids.


Adrenocorticoid Hormones
The adrenocorticoid hormones are products of the adrenal glands.
The most important mineralcorticoid is aldosterone, which regulates the
reabsorption of sodium and chloride ions in the kidney tubules and increases
the loss of potassium ions.Aldosterone is secreted when blood sodium ion
levels are too low to cause the kidney to retain sodium ions. If sodium
levels are elevated, aldosterone is not secreted, so that some sodium
will be lost in the urine. Aldosterone also controls swelling in the tissues.
Cortisol, the most important glucocortinoid, has the function of increasing
glucose and glycogen concentrations in the body. These reactions are
completed in the liver by taking fatty acids from lipid storage cells and
amino acids from body proteins to make glucose and glycogen.
In addition, cortisol is elevated in the circulation with cytokine mediated
(IL1, IL1, TNFα) inflammatory reaction, called the systemic inflammatory
response syndrome. Its ketone derivative, cortisone, has the ability
to relieve inflammatory effects. Cortisone or similar synthetic derivatives
such as prednisolone are used to treat inflammatory diseases, rheumatoid
arthritis, and bronchial asthma. There are many side effects with the use
of cortisone drugs, such as bone resorption, so there use must be
monitored carefully.
Hormone Receptors
Steroid hormone receptors are found on the plasma membrane, in the cytosol and also in the nucleus of target cells. They are generally intracellular receptors (typically cytoplasmic) and initiate signal transduction for steroid hormones which lead to changes in gene expression over a time period of hours to days. The best studied steroid hormone receptors are members of the nuclear receptor subfamily 3 (NR3) that include receptors for estrogen (group NR3A)[1] and 3-ketosteroids (group NR3C).[2] In addition to nuclear receptors, several G protein-coupled receptors and ion channels act as cell surface receptors for certain steroid hormones.
Steroid Hormone Receptors and their Response Elements
Steroid hormone receptors are proteins that have a binding site for a particular steroid molecule. Their response elements are DNA sequences that are bound by the complex of the steroid bound to its Steroid receptor.
The response element is part of the promoter of a gene. Binding by the receptor activates or represses, as the case may be, the gene controlled by that promoter.
It is through this mechanism that steroid hormones turn genes on (or off).
http://users.rcn.com/jkimball.ma.ultranet/BiologyPages/S/Sigler.jpg
{kind=link}
The glucocorticoid receptor, like all steroid hormone receptors, is a zinc-finger transcription factor; the zinc atoms are the four yellow spheres. Each is attached to four cysteines.
For a steroid hormone to regulate (turn on or off) gene transcription, its receptor must:
- bind to the hormone (cortisol in the case of the glucocorticoid receptor)
- bind to a second copy of itself to form a homodimer
- be in the nucleus, moving from the cytosol if necessary
- bind to its response element
- bind to other protein cofactors
Each of these functions depend upon a particular region of the protein (e.g., the zinc fingers for binding DNA).
Each of these functions depend upon a particular region of the protein (e.g., the zinc fingers for binding DNA). Mutations in any one region may upset the function of that region without necessarily interfering with other functions of the receptor.
Positive and Negative Response Elements
Some of the hundreds of glucocorticoid response elements in the human genome activate gene transcription when bound by the hormone/receptor complex. Others inhibit gene transcription when bound by the hormone/receptor complex.
Example: When the stress hormone cortisol — bound to its receptor — enters the nucleus of a liver cell, the complex binds to the positive response elements of the many genes needed for gluconeogenesis — the conversion of protein and fat into glucose resulting in a rise in the level of blood sugar.
the negative response element of the insulin receptor gene thus diminishing the ability of the cells to remove glucose from the blood. (This hyperglycemic effect is enhanced by the binding of the cortisol/receptor complex to a negative response element in the beta cells of the pancreas thus reducing the production of insulin.)
Note that every type of cell in the body contains the same response elements in its genome. What determines if a given cell responds to the arrival of a hormone depends on the presence of the hormone’s receptor in the cell.
The Nuclear Receptor Superfamily

http://users.rcn.com/jkimball.ma.ultranet/BiologyPages/R/Retinoids.png
{kind=link}
The zinc-finger proteins that serve as receptors for glucocorticoids and progesterone are members of a large family of similar proteins that serve as receptors for a variety of small, hydrophobic molecules. These include:
- other steroid hormones like
- the mineralocorticoid aldosterone
- estrogens
- the thyroid hormone, T3
- calcitriol, the active form of vitamin D
- retinoids: vitamin A (retinol) and its relatives
- retinal
- retinoic acid (tretinoin — also available as the drug Retin-A®); and its isomer
- isotretinoin (sold as Accutane® for the treatment of acne).
- bile acids
- fatty acids.
These bind members of the superfamily called peroxisome-proliferator-activated receptors (PPARs). They got their name from their initial discovery as the receptors for
- drugs that increase the number and size of peroxisomes in cells.
In every case, the receptors consist of at least
- three functional modules or domains.
From N-terminal to C-terminal, these are:
- a domain needed
- the zinc-finger domain needed for DNA binding (to the response element)
- the domain responsible for binding the particular hormone as well as the second unit of the dimer.
- for the receptor to activate the promoters of the genes being controlled
Schematic diagram of type II zinc finger proteins characteristic of the DNA-binding domain structure of members of the steroid hormone receptor superfamily. Zinc fingers are common features of many transcription factors, allowing proteins to bind to DNA. Each circle represents one amino acid. The CI zinc finger interacts specifically with five base pairs of DNA and determines the DNA sequence recognized by the particular steroid receptor. The three shaded amino acids indicated by the arrows in the knuckle of the CI zinc finger are in the “P box” that allows HRE sequence discrimination between the GR and ERα. The vertically striped aa within the knuckle of the CII zinc finger constitutes the “D box” that is important for dimerization and contacts with the DNA phosphate backbone. Adapted from Tsai M-J, O’Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 1994;63:451-483; Gronemeyer H. Transcription activation by estrogen and progesterone receptors. Annu Rev Genet 1991;25:89-123.

Cytoskeleton and Cell Membrane Physiology
http://pharmaceuticalinnovation.com/10/28/2014/larryhbern/Cytoskeleton_
and_Cell_Membrane_Physiology
Definition and Function
The cytoskeleton is a series of intercellular proteins that help a cell with
- shape,
- support, and
- movement.
Cytoskeleton has three main structural components:
- microfilaments,
- intermediate filaments, and
- movement
The cytoskeleton mediates movement by
- helping the cell move in its environment and
- mediating the movement of the cell’s components.
Thereby it provides an important structural framework for the cell –
- the framework for the movement of organelles, contiguous with the cell membrane, around the cytoplasm. By the activity of
- the network of protein microfilaments, intermediate filaments, and microtubules.
The structural framework supports cell function as follows:
Cell shape. For cells without cell walls, the cytoskeleton determines the shape of the cell. This is one of the functions of the intermediate filaments.
Cell movement. The dynamic collection of microfilaments and microtubles can be continually in the process of assembly and disassembly, resulting in forces that move the cell. There can also be sliding motions of these structures. Audesirk and Audesirk give examples of white blood cells “crawling” and the migration and shape changes of cells during the development of multicellular organisms.
Organelle movement. Microtubules and microfilaments can help move organelles from place to place in the cell. In endocytosis a vesicle formed engulfs a particle abutting the cell. Microfilaments then attach to the vesicle and pull it into the cell. Much of the complex synthesis and distribution function of the endoplasmic reticulum and the Golgi complex makes use of transport vescicles, associated with the cytoskeleton.
Cell division. During cell division, microtubules accomplish the movement of the chromosones to the daughter nucleus. Also, a ring of microfilaments helps divide two developing cells by constricting the central region between the cells (fission).
References:
Hickman, et al. Ch 4 Hickman, Cleveland P., Roberts, Larry S., and Larson, Allan, Integrated Principles of Zoology, 9th. Ed., Wm C. Brown, 1995.
Audesirk & Audesirk Ch 6 Audesirk, Teresa and Audesirk, Gerald, Biology, Life on Earth, 5th Ed., Prentice-Hall, 1999.
http://hyperphysics.phy-astr.gsu.edu/hbase/biology/bioref.html#c1
http://hyperphysics.phy-astr.gsu.edu/hbase/biology/cytoskel.html
Chapter 3. Cell Signaling
Introduction to Signaling
Larry H. Bernstein, MD, FCAP
We have laid down a basic structure and foundation for the remaining presentations. It was essential to begin with the genome, which changed the course of teaching of biology and medicine in the 20th century, and introduced a central dogma of translation by transcription. Nevertheless, there were significant inconsistencies and unanswered questions entering the twenty first century, accompanied by vast improvements in technical advances to clarify these issues. We have covered carbohydrate, protein, and lipid metabolism, which function in concert with the development of cellular structure, organ system development, and physiology. To be sure, the progress in the study of the microscopic and particulate can’t be divorced from the observation of the whole. We were left in the not so distant past with the impression of the Sufi story of the elephant and the three blind men, who one at a time held the tail, the trunk, and the ear, each proclaiming that it was the elephant.
I introduce here a story from the Brazilian biochemist, Jose Eduardo des Salles Rosalino, on a formative experience he had with the Nobelist, Luis Leloir.
Just at the beginning, when phosphorylation of proteins is presented, I assume you must mention that some proteins are activated by phosphorylation. This is fundamental in order to present self –organization reflex upon fast regulatory mechanisms. Even from an historical point of view. The first observation arrived from a sample due to be studied on the following day of glycogen synthetase. It was unintended left overnight out of the refrigerator. The result was it has changed from active form of the previous day to a non-active form. The story could have being finished here, if the researcher did not decide to spent this day increasing substrate levels (it could be a simple case of denaturation of proteins that changes its conformation despite the same order of amino acids). He kept on trying and found restoration of maximal activity. This assay was repeated with glycogen phosphorylase and the result was the opposite – it increases its activity. This led to the discovery
- of cAMP activated protein kinase and
- the assembly of a very complex system in the glycogen granule
- that is not a simple carbohydrate polymer.
Instead, it has several proteins assembled and
- preserves the capacity to receive from a single event (rise in cAMP)
- two opposing signals with maximal efficiency,
- stops glycogen synthesis,
- as long as levels of glucose 6 phosphate are low
- and increases glycogen phosphorylation as long as AMP levels are high).
I did everything I was able to do by the end of 1970 in order to repeat the assays with PK I, PKII and PKIII of M. Rouxii and using the Sutherland route to cAMP failed in this case. I then asked Leloir to suggest to my chief (SP) the idea of AA, AB, BB subunits as was observed in lactic dehydrogenase (tetramer) indicating this as hisidea. The reason was my “chief”(SP) more than once, had said to me: “Leave these great ideas for the Houssay, Leloir etc…We must do our career with small things.” However, as she also had a faulty ability for recollection she also used to arrive some time later, with the very same idea but in that case, as her idea.
Leloir, said to me: I will not offer your interpretation to her as mine. I think it is not phosphorylation, however I think it is glycosylation that explains the changes in the isoenzymes with the same molecular weight preserved. This dialogue explains why during the reading and discussing “What is life” with him he asked me if as a biochemist in exile, talking to another biochemist, I expressed myself fully. I had considered that Schrödinger would not have confronted Darlington & Haldane because he was in U.K. in exile. This might explain why Leloir could have answered a bad telephone call from P. Boyer, Editor of The Enzymes, in a way that suggested that the pattern could be of covalent changes over a protein. Our FEBS and Eur J. Biochemistry papers on pyruvate kinase of M. Rouxii is wrongly quoted in this way on his review about pyruvate kinase of that year (1971).
Another aspect I think you must call attention to the following. Show in detail with different colors what carbons belongs to CoA, a huge molecule in comparison with the single two carbons of acetate that will produce the enormous jump in energy yield
- in comparison with anaerobic glycolysis.
The idea is
- how much must have been spent in DNA sequences to build that molecule in order to use only two atoms of carbon.
Very limited aspects of biology could be explained in this way. In case we follow an alternative way of thinking, it becomes clearer that proteins were made more stable by interaction with other molecules (great and small). Afterwards, it’s rather easy to understand how the stability of protein-RNA complexes where transmitted to RNA (vibrational +solvational reactivity stability pair of conformational energy).
Millions of years later, or as soon as, the information of interaction leading to activity and regulation could be found in RNA, proteins like reverse transcriptase move this information to a more stable form (DNA). In this way it is easier to understand the use of CoA to make two carbon molecules more reactive.
The discussions that follow are concerned with protein interactions and signaling.
3.1 Signaling and Signaling Pathways
Larry H. Bernstein, MD, FCAP
3.2 Signaling Transduction Tutorial
Larry H. Bernstein, MD, FCAP
3.3 Selected References to Signaling and Metabolic Pathways in Leaders in Pharmaceutical Intelligence
Larry H. Bernstein, MD, FCAP
3.4 Integrins, Cadherins, Signaling and the Cytoskeleton
Larry H. Bernstein, MD, FCAP
3.5 Complex Models of Signaling: Therapeutic Implications
Larry H. Bernstein, MD, FCAP
3.6 Functional Correlates of Signaling Pathways
Larry H. Bernstein, MD, FCAP
Summary of Signaling and Signaling Pathways
Larry H Bernstein, MD, FCAP
In the imtroduction to this series of discussions I pointed out JEDS Rosalino’s observation about the construction of a complex molecule of acetyl coenzyme A, and the amount of genetic coding that had to go into it. Furthermore, he observes – Millions of years later, or as soon as, the information of interaction leading to activity and regulation could be found in RNA, proteins like reverse transcriptase move this information to a more stable form (DNA). In this way it is easier to understand the use of CoA to make two carbon molecules more reactive.

In the tutorial that follows we find support for the view that mechanisms and examples from the current literature, which give insight into the developments in cell metabolism, are achieving a separation from inconsistent views introduced by the classical model of molecular biology and genomics, toward a more functional cellular dynamics that is not dependent on the classic view. The classical view fits a rigid framework that is to genomics and metabolomics as Mendelian genetics if to multidimentional, multifactorial genetics. The inherent difficulty lies in two places:
- Interactions between differently weighted determinants
- A large part of the genome is concerned with regulatory function, not expression of the code
The goal of the tutorial was to achieve an understanding of how cell signaling occurs in a cell. Completion of the tutorial would provide
- a basic understanding signal transduction and
- the role of phosphorylation in signal transduction.

In addition – detailed knowledge of –
- the role of Tyrosine kinases and
- G protein-coupled receptors in cell signaling.



We are constantly receiving and interpreting signals from our environment, which can come
- in the form of light, heat, odors, touch or sound.
The cells of our bodies are also
- constantly receiving signals from other cells.
These signals are important to
- keep cells alive and functioning as well as
- to stimulate important events such as
- cell division and differentiation.
Signals are most often chemicals that can be found
- in the extracellular fluid around cells.
These chemicals can come
- from distant locations in the body (endocrine signaling by hormones), from
- nearby cells (paracrine signaling) or can even
- be secreted by the same cell (autocrine signaling).

Signaling molecules may trigger any number of cellular responses, including
- changing the metabolism of the cell receiving the signal or
- result in a change in gene expression (transcription) within the nucleus of the cell or both.

To which I would now add..
- result in either an inhibitory or a stimulatory effect
The three stages of cell signaling are:
Cell signaling can be divided into 3 stages:
Reception: A cell detects a signaling molecule from the outside of the cell.
Transduction: When the signaling molecule binds the receptor it changes the receptor protein in some way. This change initiates the process of transduction. Signal transduction is usually a pathway of several steps. Each relay molecule in the signal transduction pathway changes the next molecule in the pathway.
Response: Finally, the signal triggers a specific cellular response.

http://www.hartnell.edu/tutorials/biology/images/signaltransduction_simple.jpg
{kind=link}
The initiation is depicted as follows:
Signal Transduction – ligand binds to surface receptor
Membrane receptors function by binding the signal molecule (ligand) and causing the production of a second signal (also known as a second messenger) that then causes a cellular response. These types of receptors transmit information from the extracellular environment to the inside of the cell.
- by changing shape or
- by joining with another protein
- once a specific ligand binds to it.
Examples of membrane receptors include
- G Protein-Coupled Receptors and

- Receptor Tyrosine Kinases.

http://www.hartnell.edu/tutorials/biology/images/membrane_receptor_tk.jpg
{kind=link}
Intracellular receptors are found inside the cell, either in the cytopolasm or in the nucleus of the target cell (the cell receiving the signal).
Note that though change in gene expression is stated, the change in gene expression does not here imply a change in the genetic information – such as – mutation. That does not have to be the case in the normal homeostatic case.
This point is the differentiating case between what JEDS Roselino has referred as
- a fast, adaptive reaction, that is the feature of protein molecules, and distinguishes this interaction from
- a one-to-one transcription of the genetic code.
The rate of transcription can be controlled, or it can be blocked. This is in large part in response to the metabolites in the immediate interstitium.
This might only be
- a change in the rate of a transcription or a suppression of expression through RNA.
- Or through a conformational change in an enzyme

Since signaling systems need to be
- responsive to small concentrations of chemical signals and act quickly,
- cells often use a multi-step pathway that transmits the signal quickly,
- while amplifying the signal to numerous molecules at each step.
Signal transduction pathways are shown (simplified):

Signal transduction occurs when an
- extracellular signaling molecule activates a specific receptor located on the cell surface or inside the cell.
- In turn, this receptor triggers a biochemical chain of events inside the cell, creating a response.
- Depending on the cell, the response alters the cell’s metabolism, shape, gene expression, or ability to divide.
- The signal can be amplified at any step. Thus, one signaling molecule can cause many responses.
In 1970, Martin Rodbell examined the effects of glucagon on a rat’s liver cell membrane receptor. He noted that guanosine triphosphate disassociated glucagon from this receptor and stimulated the G-protein, which strongly influenced the cell’s metabolism. Thus, he deduced that the G-protein is a transducer that accepts glucagon molecules and affects the cell. For this, he shared the 1994 Nobel Prize in Physiology or Medicine with Alfred G. Gilman.

In 2007, a total of 48,377 scientific papers—including 11,211 e-review papers—were published on the subject. The term first appeared in a paper’s title in 1979. Widespread use of the term has been traced to a 1980 review article by Rodbell: Research papers focusing on signal transduction first appeared in large numbers in the late 1980s and early 1990s.
Signal transduction involves the binding of extracellular signaling molecules and ligands to cell-surface receptors that trigger events inside the cell. The combination of messenger with receptor causes a change in the conformation of the receptor, known as receptor activation.
This activation is always the initial step (the cause) leading to the cell’s ultimate responses (effect) to the messenger. Despite the myriad of these ultimate responses, they are all directly due to changes in particular cell proteins. Intracellular signaling cascades can be started through cell-substratum interactions; examples are the integrin that binds ligands in the extracellular matrix and steroids.

Most steroid hormones have receptors within the cytoplasm and act by stimulating the binding of their receptors to the promoter region of steroid-responsive genes.

Various environmental stimuli exist that initiate signal transmission processes in multicellular organisms; examples include photons hitting cells in the retina of the eye, and odorants binding to odorant receptors in the nasal epithelium. Certain microbial molecules, such as viral nucleotides and protein antigens, can elicit an immune system response against invading pathogens mediated by signal transduction processes. This may occur independent of signal transduction stimulation by other molecules, as is the case for the toll-like receptor. It may occur with help from stimulatory molecules located at the cell surface of other cells, as with T-cell receptor signaling. Receptors can be roughly divided into two major classes: intracellular receptors and extracellular receptors.
Signal transduction cascades amplify the signal output

G protein-coupled receptors (GPCRs) are a family of integral transmembrane proteins that possess seven transmembrane domains and are linked to a heterotrimeric G protein. Many receptors are in this family, including adrenergic receptors and chemokine receptors.
Arrestin binding to active GPCR kinase (GRK)-phosphorylated GPCRs blocks G protein coupling


Signal transduction by a GPCR begins with an inactive G protein coupled to the receptor; it exists as a heterotrimer consisting of Gα, Gβ, and Gγ. Once the GPCR recognizes a ligand, the conformation of the receptor changes to activate the G protein, causing Gα to bind a molecule of GTP and dissociate from the other two G-protein subunits.
The dissociation exposes sites on the subunits that can interact with other molecules. The activated G protein subunits detach from the receptor and initiate signaling from many downstream effector proteins such as phospholipases and ion channels, the latter permitting the release of second messenger molecules.
Receptor tyrosine kinases (RTKs) are transmembrane proteins with an intracellular kinase domain and an extracellular domain that binds ligands; examples include growth factor receptors such as the insulin receptor.

To perform signal transduction, RTKs need to form dimers in the plasma membrane; the dimer is stabilized by ligands binding to the receptor.

The interaction between the cytoplasmic domains stimulates the autophosphorylation of tyrosines within the domains of the RTKs, causing conformational changes.

Subsequent to this, the receptors’ kinase domains are activated, initiating phosphorylation signaling cascades of downstream cytoplasmic molecules that facilitate various cellular processes such as cell differentiation and metabolism.

As is the case with GPCRs, proteins that bind GTP play a major role in signal transduction from the activated RTK into the cell. In this case, the G proteins are
- members of the Ras, Rho, and Raf families, referred to collectively as small G proteins.
They act as molecular switches usually
- tethered to membranes by isoprenyl groups linked to their carboxyl ends.
Upon activation, they assign proteins to specific membrane subdomains where they participate in signaling. Activated RTKs in turn activate
- small G proteins that activate guanine nucleotide exchange factors such as SOS1.
Once activated, these exchange factors can activate more small G proteins, thus
- amplifying the receptor’s initial signal.
The mutation of certain RTK genes, as with that of GPCRs, can result in the expression of receptors that exist in a constitutively activate state; such mutated genes may act as oncogenes.
Integrin

Integrin-mediated signal transduction
An overview of integrin-mediated signal transduction, adapted from Hehlgens et al. (2007).
Integrins are produced by a wide variety of cells; they play a role in
- cell attachment to other cells and the extracellular matrix and
- in the transduction of signals from extracellular matrix components such as fibronectin and collagen.
Ligand binding to the extracellular domain of integrins
- changes the protein’s conformation,
- clustering it at the cell membrane to
- initiate signal transduction.
Integrins lack kinase activity; hence, integrin-mediated signal transduction is achieved through a variety of intracellular protein kinases and adaptor molecules, the main coordinator being integrin-linked kinase.
As shown in the picture, cooperative integrin-RTK signaling determines the
- timing of cellular survival,
- apoptosis,
- proliferation, and
- differentiation.


ion channel
A ligand-gated ion channel, upon binding with a ligand, changes conformation
- to open a channel in the cell membrane
- through which ions relaying signals can pass.
An example of this mechanism is found in the receiving cell of a neural synapse. The influx of ions that occurs in response to the opening of these channels
- induces action potentials, such as those that travel along nerves,
- by depolarizing the membrane of post-synaptic cells,
- resulting in the opening of voltage-gated ion channels.

An example of an ion allowed into the cell during a ligand-gated ion channel opening is Ca2+;
- it acts as a second messenger
- initiating signal transduction cascades and
- altering the physiology of the responding cell.
This results in amplification of the synapse response between synaptic cells
- by remodelling the dendritic spines involved in the synapse.
In eukaryotic cells, most intracellular proteins activated by a ligand/receptor interaction possess an enzymatic activity; examples include tyrosine kinase and phosphatases. Some of them create second messengers such as cyclic AMP and IP3,


- the latter controlling the release of intracellular calcium stores into the cytoplasm.
Many adaptor proteins and enzymes activated as part of signal transduction possess specialized protein domains that bind to specific secondary messenger molecules. For example,
- calcium ions bind to the EF hand domains of calmodulin,
- allowing it to bind and activate calmodulin-dependent kinase.

PIP3 and other phosphoinositides do the same thing to the Pleckstrin homology domains of proteins such as the kinase protein AKT.
Signals can be generated within organelles, such as chloroplasts and mitochondria, modulating the nuclear
gene expression in a process called retrograde signaling.
Recently, integrative genomics approaches, in which correlation analysis has been applied on transcript and metabolite profiling data of Arabidopsis thaliana, revealed the identification of metabolites which are putatively acting as mediators of nuclear gene expression.
Related articles
- Systems Biology Approach Reveals Genome to Phenome Correlation in Type 2 Diabetes (plosone.org)
- Gene Expression and Thiopurine Metabolite Profiling in Inflammatory Bowel Disease – Novel Clues to Drug Targets and Disease Mechanisms? (plosone.org)
- Activation of the Jasmonic Acid Plant Defence Pathway Alters the Composition of Rhizosphere
Nutrients 2014, 6, 3245-3258; http://dx.doi.org:/10.3390/nu6083245
Omega-3 (ω-3) fatty acids are one of the two main families of long chain polyunsaturated fatty acids (PUFA). The main omega-3 fatty acids in the mammalian body are
- α-linolenic acid (ALA), docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA).
Central nervous tissues of vertebrates are characterized by a high concentration of omega-3 fatty acids. Moreover, in the human brain,
- DHA is considered as the main structural omega-3 fatty acid, which comprises about 40% of the PUFAs in total.
DHA deficiency may be the cause of many disorders such as depression, inability to concentrate, excessive mood swings, anxiety, cardiovascular disease, type 2 diabetes, dry skin and so on.
On the other hand,
- zinc is the most abundant trace metal in the human brain.
There are many scientific studies linking zinc, especially
- excess amounts of free zinc, to cellular death.
Neurodegenerative diseases, such as Alzheimer’s disease, are characterized by altered zinc metabolism. Both animal model studies and human cell culture studies have shown a possible link between
- omega-3 fatty acids, zinc transporter levels and
- free zinc availability at cellular levels.
Many other studies have also suggested a possible
- omega-3 and zinc effect on neurodegeneration and cellular death.
Therefore, in this review, we will examine
- the effect of omega-3 fatty acids on zinc transporters and
- the importance of free zinc for human neuronal cells.
Moreover, we will evaluate the collective understanding of
- mechanism(s) for the interaction of these elements in neuronal research and their
- significance for the diagnosis and treatment of neurodegeneration.
Epidemiological studies have linked high intake of fish and shellfish as part of the daily diet to
- reduction of the incidence and/or severity of Alzheimer’s disease (AD) and senile mental decline in
Omega-3 fatty acids are one of the two main families of a broader group of fatty acids referred to as polyunsaturated fatty acids (PUFAs). The other main family of PUFAs encompasses the omega-6 fatty acids. In general, PUFAs are essential in many biochemical events, especially in early post-natal development processes such as
- cellular differentiation,
- photoreceptor membrane biogenesis and
- active synaptogenesis.
Despite the significance of these
two families, mammals cannot synthesize PUFA de novo, so they must be ingested from dietary sources. Though belonging to the same family, both
- omega-3 and omega-6 fatty acids are metabolically and functionally distinct and have
- opposing physiological effects. In the human body,
- high concentrations of omega-6 fatty acids are known to increase the formation of prostaglandins and
- thereby increase inflammatory processes [10].
the reverse process can be seen with increased omega-3 fatty acids in the body.
Many other factors, such as
- thromboxane A2 (TXA2),
- leukotriene
- B4 (LTB4),
- IL-1,
- IL-6,
- tumor necrosis factor (TNF) and
- C-reactive protein,
which are implicated in various health conditions, have been shown to be increased with high omega-6 fatty acids but decreased with omega-3 fatty acids in the human body.
Dietary fatty acids have been identified as protective factors in coronary heart disease, and PUFA levels are known to play a critical role in
- immune responses,
- gene expression and
- intercellular communications.
omega-3 fatty acids are known to be vital in
- the prevention of fatal ventricular arrhythmias, and
- are also known to reduce thrombus formation propensity by decreasing platelet aggregation, blood viscosity and fibrinogen levels
Since omega-3 fatty acids are prevalent in the nervous system, it seems logical that a deficiency may result in neuronal problems, and this is indeed what has been identified and reported.
In another study conducted with individuals of 65 years of age or older (n = 6158), it was found that
- only high fish consumption, but
- not dietary omega-3 acid intake,
- had a protective effect on cognitive decline
In 2005, based on a meta-analysis of the available epidemiology and preclinical studies, clinical trials were conducted to assess the effects of omega-3 fatty acids on cognitive protection. Four of the trials completed have shown
a protective effect of omega-3 fatty acids only among those with mild cognitive impairment conditions.
A trial of subjects with mild memory complaints demonstrated
- an improvement with 900 mg of DHA.
We review key findings on
- the effect of the omega-3 fatty acid DHA on zinc transporters and the
- importance of free zinc to human neuronal cells.
DHA is the most abundant fatty acid in neural membranes, imparting appropriate
- fluidity and other properties,
and is thus considered as the most important fatty acid in neuronal studies. DHA is well conserved throughout the mammalian species despite their dietary differences. It is mainly concentrated
- in membrane phospholipids at synapses and
- in retinal photoreceptors and
- also in the testis and sperm.
In adult rats’ brain, DHA comprises approximately
- 17% of the total fatty acid weight, and
- in the retina it is as high as 33%.
DHA is believed to have played a major role in the evolution of the modern human –
- in particular the well-developed brain.
Premature babies fed on DHA-rich formula show improvements in vocabulary and motor performance.
Analysis of human cadaver brains have shown that
- people with AD have less DHA in their frontal lobe
- and hippocampus compared with unaffected individuals
Furthermore, studies in mice have increased support for the
- protective role of omega-3 fatty acids.
Mice administrated with a dietary intake of DHA showed
- an increase in DHA levels in the hippocampus.
Errors in memory were decreased in these mice and they demonstrated
- reduced peroxide and free radical levels,
- suggesting a role in antioxidant defense.
Another study conducted with a Tg2576 mouse model of AD demonstrated that dietary
- DHA supplementation had a protective effect against reduction in
- drebrin (actin associated protein), elevated oxidation, and to some extent, apoptosis via
- decreased caspase activity.
Zinc
Zinc is a trace element, which is indispensable for life, and it is the second most abundant trace element in the body. It is known to be related to
- growth,
- development,
- differentiation,
- immune response,
- receptor activity,
- DNA synthesis,
- gene expression,
- neuro-transmission,
- enzymatic catalysis,
- hormonal storage and release,
- tissue repair,
- memory,
- the visual process
and many other cellular functions. Moreover, the indispensability of zinc to the body can be discussed in many other aspects, as
- a component of over 300 different enzymes
- an integral component of a metallothioneins
- a gene regulatory protein.
Approximately 3% of all proteins contain
- zinc binding motifs .
The broad biological functionality of zinc is thought to be due to its stable chemical and physical properties. Zinc is considered to have three different functions in enzymes;
- catalytic,
- coactive and
Indeed, it is the only metal found in all six different subclasses
of enzymes. The essential nature of zinc to the human body can be clearly displayed by studying the wide range of pathological effects of zinc deficiency. Anorexia, embryonic and post-natal growth retardation, alopecia, skin lesions, difficulties in wound healing, increased hemorrhage tendency and severe reproductive abnormalities, emotional instability, irritability and depression are just some of the detrimental effects of zinc deficiency.
Proper development and function of the central nervous system (CNS) is highly dependent on zinc levels. In the mammalian organs, zinc is mainly concentrated in the brain at around 150 μm. However, free zinc in the mammalian brain is calculated to be around 10 to 20 nm and the rest exists in either protein-, enzyme- or nucleotide bound form. The brain and zinc relationship is thought to be mediated
- through glutamate receptors, and
- it inhibits excitatory and inhibitory receptors.
Vesicular localization of zinc in pre-synaptic terminals is a characteristic feature of brain-localized zinc, and
- its release is dependent on neural activity.
Retardation of the growth and development of CNS tissues have been linked to low zinc levels. Peripheral neuropathy, spina bifida, hydrocephalus, anencephalus, epilepsy and Pick’s disease have been linked to zinc deficiency. However, the body cannot tolerate excessive amounts of zinc.
The relationship between zinc and neurodegeneration, specifically AD, has been interpreted in several ways. One study has proposed that β-amyloid has a greater propensity to
- form insoluble amyloid in the presence of
- high physiological levels of zinc.
Insoluble amyloid is thought to
- aggregate to form plaques,
which is a main pathological feature of AD. Further studies have shown that
- chelation of zinc ions can deform and disaggregate plaques.
In AD, the most prominent injuries are found in
- hippocampal pyramidal neurons, acetylcholine-containing neurons in the basal forebrain, and in
- somatostatin-containing neurons in the forebrain.
All of these neurons are known to favor
- rapid and direct entry of zinc in high concentration
- leaving neurons frequently exposed to high dosages of zinc.
This is thought to promote neuronal cell damage through oxidative stress and mitochondrial dysfunction. Excessive levels of zinc are also capable of
- inhibiting Ca2+ and Na+ voltage gated channels
- and up-regulating the cellular levels of reactive oxygen species (ROS).
High levels of zinc are found in Alzheimer’s brains indicating a possible zinc related neurodegeneration. A study conducted with mouse neuronal cells has shown that even a 24-h exposure to high levels of zinc (40 μm) is sufficient to degenerate cells.
If the human diet is deficient in zinc, the body
- efficiently conserves zinc at the tissue level by compensating other cellular mechanisms
to delay the dietary deficiency effects of zinc. These include reduction of cellular growth rate and zinc excretion levels, and
- redistribution of available zinc to more zinc dependent cells or organs.
A novel method of measuring metallothionein (MT) levels was introduced as a biomarker for the
- assessment of the zinc status of individuals and populations.
In humans, erythrocyte metallothionein (E-MT) levels may be considered as an indicator of zinc depletion and repletion, as E-MT levels are sensitive to dietary zinc intake. It should be noted here that MT plays an important role in zinc homeostasis by acting
- as a target for zinc ion binding and thus
- assisting in the trafficking of zinc ions through the cell,
- which may be similar to that of zinc transporters
Zinc Transporters
Deficient or excess amounts of zinc in the body can be catastrophic to the integrity of cellular biochemical and biological systems. The gastrointestinal system controls the absorption, excretion and the distribution of zinc, although the hydrophilic and high-charge molecular characteristics of zinc are not favorable for passive diffusion across the cell membranes. Zinc movement is known to occur
- via intermembrane proteins and zinc transporter (ZnT) proteins
These transporters are mainly categorized under two metal transporter families; Zip (ZRT, IRT like proteins) and CDF/ZnT (Cation Diffusion Facilitator), also known as SLC (Solute Linked Carrier) gene families: Zip (SLC-39) and ZnT (SLC-30). More than 20 zinc transporters have been identified and characterized over the last two decades (14 Zips and 8 ZnTs).
Members of the SLC39 family have been identified as the putative facilitators of zinc influx into the cytosol, either from the extracellular environment or from intracellular compartments (Figure 1).
The identification of this transporter family was a result of gene sequencing of known Zip1 protein transporters in plants, yeast and human cells. In contrast to the SLC39 family, the SLC30 family facilitates the opposite process, namely zinc efflux from the cytosol to the extracellular environment or into luminal compartments such as secretory granules, endosomes and synaptic vesicles; thus decreasing intracellular zinc availability (Figure 1). ZnT3 is the most important in the brain where
- it is responsible for the transport of zinc into the synaptic vesicles of
- glutamatergic neurons in the hippocampus and neocortex,
Figure 1: Subcellular localization and direction of transport of the zinc transporter families, ZnT and ZIP. Arrows show the direction of zinc mobilization for the ZnT (green) and ZIP (red) proteins. A net gain in cytosolic zinc is achieved by the transportation of zinc from the extracellular region and organelles such as the endoplasmic reticulum (ER) and Golgi apparatus by the ZIP transporters. Cytosolic zinc is mobilized into early secretory compartments such as the ER and Golgi apparatus by the ZnT transporters. Figures were produced using Servier Medical Art,http://www.servier.com/. http://www.hindawi.com/journals/jnme/2012/173712.fig.001.jpg
{kind=link}
Figure 2: Early zinc signaling (EZS) and late zinc signaling (LZS). EZS involves transcription-independent mechanisms where an extracellular stimulus directly induces an increase in zinc levels within several minutes by releasing zinc from intracellular stores (e.g., endoplasmic reticulum). LSZ is induced several hours after an external stimulus and is dependent on transcriptional changes in zinc transporter expression. Components of this figure were produced using Servier Medical Art
http://www.servier.com/ and adapted from Fukada et al. [30].
omega-3 fatty acids in the mammalian body are
- α-linolenic acid (ALA),
- docosahexenoic acid (DHA) and
- eicosapentaenoic acid (EPA).
In general, seafood is rich in omega-3 fatty acids, more specifically DHA and EPA (Table 1). Thus far, there are nine separate epidemiological studies that suggest a possible link between
- increased fish consumption and reduced risk of AD
- and eight out of ten studies have reported a link between higher blood omega-3 levels
DHA and Zinc Homeostasis
Many studies have identified possible associations between DHA levels, zinc homeostasis, neuroprotection and neurodegeneration. Dietary DHA deficiency resulted in
- increased zinc levels in the hippocampus and
- elevated expression of the putative zinc transporter, ZnT3, in the rat brain.
Altered zinc metabolism in neuronal cells has been linked to neurodegenerative conditions such as AD. A study conducted with transgenic mice has shown a significant link between ZnT3 transporter levels and cerebral amyloid plaque pathology. When the ZnT3 transporter was silenced in transgenic mice expressing cerebral amyloid plaque pathology,
- a significant reduction in plaque load
- and the presence of insoluble amyloid were observed.
In addition to the decrease in plaque load, ZnT3 silenced mice also exhibited a significant
- reduction in free zinc availability in the hippocampus
- and cerebral cortex.
Collectively, the findings from this study are very interesting and indicate a clear connection between
- zinc availability and amyloid plaque formation,
thus indicating a possible link to AD.
DHA supplementation has also been reported to limit the following:
- amyloid presence,
- synaptic marker loss,
- hyper-phosphorylation of Tau,
- oxidative damage and
- cognitive deficits in transgenic mouse model of AD.
In addition, studies by Stoltenberg, Flinn and colleagues report on the modulation of zinc and the effect in transgenic mouse models of AD. Given that all of these are classic pathological features of AD, and considering the limiting nature of DHA in these processes, it can be argued that DHA is a key candidate in preventing or even curing this debilitating disease.
In order to better understand the possible links and pathways of zinc and DHA with neurodegeneration, we designed a study that incorporates all three of these aspects, to study their effects at the cellular level. In this study, we were able to demonstrate a possible link between omega-3 fatty acid (DHA) concentration, zinc availability and zinc transporter expression levels in cultured human neuronal cells.
When treated with DHA over 48 h, ZnT3 levels were markedly reduced in the human neuroblastoma M17 cell line. Moreover, in the same study, we were able to propose a possible
- neuroprotective mechanism of DHA,
which we believe is exerted through
- a reduction in cellular zinc levels (through altering zinc transporter expression levels)
- that in turn inhibits apoptosis.
DHA supplemented M17 cells also showed a marked depletion of zinc uptake (up to 30%), and
- free zinc levels in the cytosol were significantly low compared to the control
This reduction in free zinc availability was specific to DHA; cells treated with EPA had no significant change in free zinc levels (unpublished data). Moreover, DHA-repleted cells had
- low levels of active caspase-3 and
- high Bcl-2 levels compared to the control treatment.
These findings are consistent with previous published data and further strengthen the possible
- correlation between zinc, DHA and neurodegeneration.
On the other hand, recent studies using ZnT3 knockout (ZnT3KO) mice have shown the importance of
- ZnT3 in memory and AD pathology.
For example, Sindreu and colleagues have used ZnT3KO mice to establish the important role of
- ZnT3 in zinc homeostasis that modulates presynaptic MAPK signaling
- required for hippocampus-dependent memory
Results from these studies indicate a possible zinc-transporter-expression-level-dependent mechanism for DHA neuroprotection.
Chapter 4. Protein Synthesis and Degradation
Introduction to Protein Synthesis and Degradation
Curator: Larry H. Bernstein, MD, FCAP
This chapter I made to follow signaling, rather than to precede it. I had already written much of the content before reorganizing the contents. The previous chapters on carbohydrate and on lipid metabolism have already provided much material on proteins and protein function, which was persuasive of the need to introduce signaling, which entails a substantial introduction to conformational changes in proteins that direct the trafficking of metabolic pathways, but more subtly uncovers an important role for microRNAs, not divorced from transcription, but involved in a non-transcriptional role. This is where the classic model of molecular biology lacked any integration with emerging metabolic concepts concerning regulation. Consequently, the science was bereft of understanding the ties between the multiple convergence of transcripts, the selective inhibition of transcriptions, and the relative balance of aerobic and anaerobic metabolism, the weight of the pentose phosphate shunt, and the utilization of available energy source for synthetic and catabolic adaptive responses.
The first subchapter serves to introduce the importance of transcription in translational science. The several subtitles that follow are intended to lay out the scope of the transcriptional activity, and also to direct attention toward the huge role of proteomics in the cell construct. As we have already seen, proteins engage with carbohydrates and with lipids in important structural and signaling processes. They are integrasl to the composition of the cytoskeleton, and also to the extracellular matrix. Many proteins are actually enzymes, carrying out the transformation of some substrate, a derivative of the food we ingest. They have a catalytic site, and they function with a cofactor – either a multivalent metal or a nucleotide.
The amino acids that go into protein synthesis include “indispensable” nutrients that are not made for use, but must be derived from animal protein, although the need is partially satisfied by plant sources. The essential amino acids are classified into well established groups. There are 20 amino acids commonly found in proteins. They are classified into the following groups based on the chemical and/or structural properties of their side chains :
- Aliphatic Amino Acids
- Cyclic Amino Acid
- AAs with Hydroxyl or Sulfur-containing side chains
- Aromatic Amino Acids
- Basic Amino Acids
- Acidic Amino Acids and their Amides
Examples include:
Alanine aliphatic hydrophobic neutral
Arginine polar hydrophilic charged (+)
Cysteine polar hydrophobic neutral
Glutamine polar hydrophilic neutral
Histidine aromatic polar hydrophilic charged (+)
Lysine polar hydrophilic charged (+)
Methionine hydrophobic neutral
Serine polar hydrophilic neutral
Tyrosine aromatic polar hydrophobic
Transcribe and Translate a Gene
- For each RNA base there is a corresponding DNA base
- Cells use the two-step process of transcription and translation to read each gene and produce the string of amino acids that makes up a protein.
- mRNA is produced in the nucleus, and is transferred to the ribosome
- mRNA uses uracil instead of thymine
- the ribosome reads the RNA sequence and makes protein
- There is a sequence combination to fit each amino acid to a three letter RNA code
- The ribosome starts at AUG (start), and it reads each codon three letters at a time
- Stop codons are UAA, UAG and UGA

http://learn.genetics.utah.edu/content/molecules/transcribe/images/TandT.png
{kind=link}

http://www.vcbio.science.ru.nl/images/cellcycle/mcell-transcription-translation_eng_zoom.gif
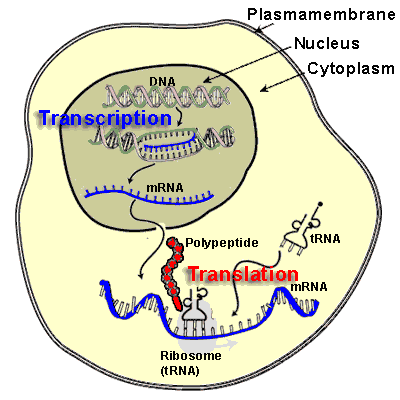{kind=link}

http://www.biologycorner.com/resources/transcription_translation.JPG
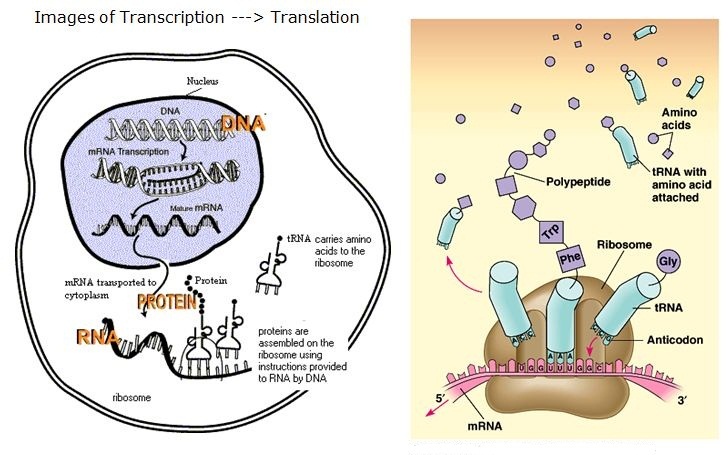{kind=link}
What about the purine inosine?
Inosine triphosphate pyrophosphatase – Pyrophosphatase that hydrolyzes the non-canonical purine nucleotides inosine triphosphate (ITP), deoxyinosine triphosphate (dITP) as well as 2′-deoxy-N-6-hydroxylaminopurine triposphate (dHAPTP) and xanthosine 5′-triphosphate (XTP) to their respective monophosphate derivatives. The enzyme does not distinguish between the deoxy- and ribose forms. Probably excludes non-canonical purines from RNA and DNA precursor pools, thus preventing their incorporation into RNA and DNA and avoiding chromosomal lesions.
Gastroenterology. 2011 Apr;140(4):1314-21. http://dx.doi.org:/10.1053/j.gastro.2010.12.038. Epub 2011 Jan 1.
Inosine triphosphate protects against ribavirin-induced adenosine triphosphate loss by adenylosuccinate synthase function.
Hitomi Y1, Cirulli ET, Fellay J, McHutchison JG, Thompson AJ, Gumbs CE, Shianna KV, Urban TJ, Goldstein DB.
Genetic variation of inosine triphosphatase (ITPA) causing an accumulation of inosine triphosphate (ITP) has been shown to protect patients against ribavirin (RBV)-induced anemia during treatment for chronic hepatitis C infection by genome-wide association study (GWAS). However, the biologic mechanism by which this occurs is unknown.
Although ITP is not used directly by human erythrocyte ATPase, it can be used for ATP biosynthesis via ADSS in place of guanosine triphosphate (GTP). With RBV challenge, erythrocyte ATP reduction was more severe in the wild-type ITPA genotype than in the hemolysis protective ITPA genotype. This difference also remains after inhibiting adenosine uptake using nitrobenzylmercaptopurine riboside (NBMPR).
ITP confers protection against RBV-induced ATP reduction by substituting for erythrocyte GTP, which is depleted by RBV, in the biosynthesis of ATP. Because patients with excess ITP appear largely protected against anemia, these results confirm that RBV-induced anemia is due primarily to the effect of the drug on GTP and consequently ATP levels in erythrocytes.
Ther Drug Monit. 2012 Aug;34(4):477-80. http://dx.doi.org:/10.1097/FTD.0b013e31825c2703.
Determination of inosine triphosphate pyrophosphatase phenotype in human red blood cells using HPLC.
Citterio-Quentin A1, Salvi JP, Boulieu R.
Thiopurine drugs, widely used in cancer chemotherapy, inflammatory bowel disease, and autoimmune hepatitis, are responsible for common adverse events. Only some of these may be explained by genetic polymorphism of thiopurine S-methyltransferase. Recent articles have reported that inosine triphosphate pyrophosphatase (ITPase) deficiency was associated with adverse drug reactions toward thiopurine drug therapy. Here, we report a weak anion exchange high-performance liquid chromatography method to determine ITPase activity in red blood cells and to investigate the relationship with the occurrence of adverse events during azathioprine therapy.
The chromatographic method reported allows the analysis of IMP, inosine diphosphate, and ITP in a single run in <12.5 minutes. The method was linear in the range 5-1500 μmole/L of IMP. Intraassay and interassay precisions were <5% for red blood cell lysates supplemented with 50, 500, and 1000 μmole/L IMP. Km and Vmax evaluated by Lineweaver-Burk plot were 677.4 μmole/L and 19.6 μmole·L·min, respectively. The frequency distribution of ITPase from 73 patients was investigated.
The method described is useful to determine the ITPase phenotype from patients on thiopurine therapy and to investigate the potential relation between ITPase deficiency and the occurrence of adverse events.
System wide analyses have underestimated protein abundances and the importance of transcription in mammals
Jingyi Jessica Li1, 2, Peter J Bickel1 and Mark D Biggin3
PeerJ 2:e270; http://dx.doi.org:/10.7717/peerj.270
Using individual measurements for 61 housekeeping proteins to rescale whole proteome data from Schwanhausser et al. (2011), we find that the median protein detected is expressed at 170,000 molecules per cell and that our corrected protein abundance estimates show a higher correlation with mRNA abundances than do the uncorrected protein data. In addition, we estimated the impact of further errors in mRNA and protein abundances using direct experimental measurements of these errors. The resulting analysis suggests that mRNA levels explain at least 56% of the differences in protein abundance for the 4,212 genes detected by Schwanhausser et al. (2011), though because one major source of error could not be estimated the true percent contribution should be higher.We also employed a second, independent strategy to determine the contribution of mRNA levels to protein expression.We show that the variance in translation rates directly measured by ribosome profiling is only 12% of that inferred by Schwanhausser et al. (2011), and that the measured and inferred translation rates correlate poorly (R2 D 0.13). Based on this, our second strategy suggests that mRNA levels explain 81% of the variance in protein levels. We also determined the percent contributions of transcription, RNA degradation, translation and protein degradation to the variance in protein abundances using both of our strategies. While the magnitudes of the two estimates vary, they both suggest that transcription plays a more important role than the earlier studies implied and translation a much smaller role. Finally, the above estimates only apply to those genes whose mRNA and protein expression was detected. Based on a detailed analysis by Hebenstreit et al. (2012), we estimat that approximately 40% of genes in a given cell within a population express no mRNA. Since there can be no translation in the ab-sence of mRNA, we argue that differences in translation rates can play no role in determining the expression levels for the 40% of genes that are non-expressed.
Related studies that reveal issues that are not part of this chapter:
- Ubiquitylation in relationship to tissue remodeling
- Post-translational modification of proteins
- Glycosylation
- Phosphorylation
- Methylation
- Nitrosylation
- Sulfation – sulfotransferases
cell-matrix communication - Acetylation and histone deacetylation (HDAC)
Connecting Protein Phosphatase to 1α (PP1α)
Acetylation complexes (such as CBP/p300 and PCAF)
Sirtuins
Rel/NF-kB Signal Transduction
Homologous Recombination Pathway of Double-Strand DNA Repair - Glycination
- cyclin dependent kinases (CDKs)
- lyase
- transferase
This year, the Lasker award for basic medical research went to Kazutoshi Mori (Kyoto University) and Peter Walter (University of California, San Francisco) for their “discoveries concerning the unfolded protein response (UPR) — an intracellular quality control system that
detects harmful misfolded proteins in the endoplasmic reticulum and signals the nucleus to carry out corrective measures.”
About UPR: Approximately a third of cellular proteins pass through the Endoplasmic Reticulum (ER) which performs stringent quality control of these proteins. All proteins need to assume the proper 3-dimensional shape in order to function properly in the harsh cellular environment. Related to this is the fact that cells are under constant stress and have to make rapid, real time decisions about survival or death.
A major indicator of stress is the accumulation of unfolded proteins within the Endoplasmic Reticulum (ER), which triggers a transcriptional cascade in order to increase the folding capacity of the ER. If the metabolic burden is too great and homeostasis cannot be achieved, the response shifts from
damage control to the induction of pro-apoptotic pathways that would ultimately cause cell death.
This response to unfolded proteins or the UPR is conserved among all eukaryotes, and dysfunction in this pathway underlies many human diseases, including Alzheimer’s, Parkinson’s, Diabetes and Cancer.
The discovery of a new class of human proteins with previously unidentified activities
In a landmark study conducted by scientists at the Scripps Research Institute, The Hong Kong University of Science and Technology, aTyr Pharma and their collaborators, a new class of human proteins has been discovered. These proteins [nearly 250], called Physiocrines belong to the aminoacyl tRNA synthetase gene family and carry out novel, diverse and distinct biological functions.
The aminoacyl tRNA synthetase gene family codes for a group of 20 ubiquitous enzymes almost all of which are part of the protein synthesis machinery. Using recombinant protein purification, deep sequencing technique, mass spectroscopy and cell based assays, the team made this discovery. The finding is significant, also because it highlights the alternate use of a gene family whose protein product normally performs catalytic activities for non-catalytic regulation of basic and complex physiological processes spanning metabolism, vascularization, stem cell biology and immunology
Muscle maintenance and regeneration – key player identified
Muscle tissue suffers from atrophy with age and its regenerative capacity also declines over time. Most molecules discovered thus far to boost tissue regeneration are also implicated in cancers. During a quest to find safer alternatives that can regenerate tissue, scientists reported that the hormone Oxytocin is required for proper muscle tissue regeneration and homeostasis and that its levels decline with age.
Oxytocin could be an alternative to hormone replacement therapy as a way to combat aging and other organ related degeneration.
Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration (June 2014)
Proc Natl Acad Sci U S A. 2014 Sep 30;111(39):14289-94. http://dx.doi.org:/10.1073/pnas.1407640111. Epub 2014 Sep 15.
Role of forkhead box protein A3 in age-associated metabolic decline.
Ma X1, Xu L1, Gavrilova O2, Mueller E3.
Aging is associated with increased adiposity and diminished thermogenesis, but the critical transcription factors influencing these metabolic changes late in life are poorly understood. We recently demonstrated that the winged helix factor forkhead box protein A3 (Foxa3) regulates the expansion of visceral adipose tissue in high-fat diet regimens; however, whether Foxa3 also contributes to the increase in adiposity and the decrease in brown fat activity observed during the normal aging process is currently unknown. Here we report that during aging, levels of Foxa3 are significantly and selectively up-regulated in brown and inguinal white fat depots, and that midage Foxa3-null mice have increased white fat browning and thermogenic capacity, decreased adipose tissue expansion, improved insulin sensitivity, and increased longevity. Foxa3 gain-of-function and loss-of-function studies in inguinal adipose depots demonstrated a cell-autonomous function for Foxa3 in white fat tissue browning. Furthermore, our analysis revealed that the mechanisms of Foxa3 modulation of brown fat gene programs involve the suppression of peroxisome proliferator activated receptor γ coactivtor 1 α (PGC1α) levels through interference with cAMP responsive element binding protein 1-mediated transcriptional regulation of the PGC1α promoter.
Asymmetric mRNA localization contributes to fidelity and sensitivity of spatially localized systems
RJ Weatheritt, TJ Gibson & MM Babu
Nature Structural & Molecular Biology 24 Aug, 2014; 21: 833–839http://dx.do.orgi:/10.1038/nsmb.2876
Although many proteins are localized after translation, asymmetric protein distribution is also achieved by translation after mRNA localization. Why are certain mRNA transported to a distal location and translated on-site? Here we undertake a systematic, genome-scale study of asymmetrically distributed protein and mRNA in mammalian cells. Our findings suggest that asymmetric protein distribution by mRNA localization enhances interaction fidelity and signaling sensitivity. Proteins synthesized at distal locations frequently contain intrinsically disordered segments. These regions are generally rich in assembly-promoting modules and are often regulated by post-translational modifications. Such proteins are tightly regulated but display distinct temporal dynamics upon stimulation with growth factors. Thus, proteins synthesized on-site may rapidly alter proteome composition and act as dynamically regulated scaffolds to promote the formation of reversible cellular assemblies. Our observations are consistent across multiple mammalian species, cell types and developmental stages, suggesting that localized translation is a recurring feature of cell signaling and regulation.
An overview of the potential advantages conferred by distal-site protein synthesis, inferred from our analysis.

Turquoise and red filled circle represents off-target and correct interaction partners, respectively. Wavy lines represent a disordered region within a distal site synthesis protein. Grey and red line in graphs represents profiles of t…
http://www.nature.com/nsmb/journal/v21/n9/carousel/nsmb.2876-F5.jpg
{kind=link}
Tweaking transcriptional programming for high quality recombinant protein production
Since overexpression of recombinant proteins in E. coli often leads to the formation of inclusion bodies, producing properly folded, soluble proteins is undoubtedly the most important end goal in a protein expression campaign. Various approaches have been devised to bypass the insolubility issues during E. coli expression and in a recent report a group of researchers discuss reprogramming the E. coli proteostasis [protein homeostasis] network to achieve high yields of soluble, functional protein. The premise of their studies is that the basal E. coli proteostasis network is insufficient, and often unable, to fold overexpressed proteins, thus clogging the folding machinery.
By overexpressing a mutant, negative-feedback deficient heat shock transcription factor [σ32 I54N] before and during overexpression of the protein of interest, reprogramming can be achieved, resulting in high yields of soluble and functional recombinant target protein. The authors explain that this method is better than simply co-expressing/over-expressing chaperones, co-chaperones, foldases or other components of the proteostasis network because reprogramming readies the folding machinery and up regulates the essential folding components beforehand thus maintaining system capability of the folding machinery.
The Heat-Shock Response Transcriptional Program Enables High-Yield and High-Quality Recombinant Protein Production in Escherichia coli (July 2014)
Unfolded proteins collapse when exposed to heat and crowded environments
Proteins are important molecules in our body and they fulfil a broad range of functions. For instance as enzymes they help to release energy from food and as muscle proteins they assist with motion. As antibodies they are involved in immune defence and as hormone receptors in signal transduction in cells. Until only recently it was assumed that all proteins take on a clearly defined three-dimensional structure – i.e. they fold in order to be able to assume these functions. Surprisingly, it has been shown that many important proteins occur as unfolded coils. Researchers seek to establish how these disordered proteins are capable at all of assuming highly complex functions.
Ben Schuler’s research group from the Institute of Biochemistry of the University of Zurich has now established that an increase in temperature leads to folded proteins collapsing and becoming smaller. Other environmental factors can trigger the same effect.
Measurements using the “molecular ruler”
“The fact that unfolded proteins shrink at higher temperatures is an indication that cell water does indeed play an important role as to the spatial organisation eventually adopted by the molecules”, comments Schuler with regard to the impact of temperature on protein structure. For their studies the biophysicists use what is known as single-molecule spectroscopy. Small colour probes in the protein enable the observation of changes with an accuracy of more than one millionth of a millimetre. With this “molecular yardstick” it is possible to measure how molecular forces impact protein structure.
With computer simulations the researchers have mimicked the behaviour of disordered proteins.
(Courtesy of Jose EDS Roselino, PhD.
MLKL compromises plasma membrane integrity
Necroptosis is implicated in many diseases and understanding this process is essential in the search for new therapies. While mixed lineage kinase domain-like (MLKL) protein has been known to be a critical component of necroptosis induction, how MLKL transduces the death signal was not clear. In a recent finding, scientists demonstrated that the full four-helical bundle domain (4HBD) in the N-terminal region of MLKL is required and sufficient to induce its oligomerization and trigger cell death.
They also found a patch of positively charged amino acids on the surface of the 4HBD that bound to phosphatidylinositol phosphates (PIPs) and allowed the recruitment of MLKL to the plasma membrane that resulted in the formation of pores consisting of MLKL proteins, due to which cells absorbed excess water causing them to explode. Detailed knowledge about how MLKL proteins create pores offers possibilities for the development of new therapeutic interventions for tolerating or preventing cell death.
MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates (May 2014)
Mitochondrial and ER proteins implicated in dementia
Mitochondria and the endoplasmic reticulum (ER) form tight structural associations that facilitate a number of cellular functions. However, the molecular mechanisms of these interactions aren’t properly understood.
A group of researchers showed that the ER protein VAPB interacted with mitochondrial protein PTPIP51 to regulate ER-mitochondria associations and that TDP-43, a protein implicated in dementia, disturbs this interaction to regulate cellular Ca2+ homeostasis. These studies point to a new pathogenic mechanism for TDP-43 and may also provide a potential new target for the development of new treatments for devastating neurological conditions like dementia.
ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nature (June 2014)
A novel strategy to improve membrane protein expression in Yeast
Membrane proteins play indispensable roles in the physiology of an organism. However, recombinant production of membrane proteins is one of the biggest hurdles facing protein biochemists today. A group of scientists in Belgium showed that,
by increasing the intracellular membrane production by interfering with a key enzymatic step of lipid synthesis,
enhanced expression of recombinant membrane proteins in yeast is achieved.
Specifically, they engineered the oleotrophic yeast, Yarrowia lipolytica, by
deleting the phosphatidic acid phosphatase, PAH1 gene,
which led to massive proliferation of endoplasmic reticulum (ER) membranes.
For all 8 tested representatives of different integral membrane protein families, they obtained enhanced protein accumulation.
An unconventional method to boost recombinant protein levels
MazF is an mRNA interferase enzyme in E.coli that functions as and degrades cellular mRNA in a targeted fashion, at the “ACA” sequence. This degradation of cellular mRNA causes a precipitous drop in cellular protein synthesis. A group of scientists at the Robert Wood Johnson Medical School in New Jersey, exploited the degeneracy of the genetic code to modify all “ACA” triplets within their gene of interest in a way that the corresponding amino acid (Threonine) remained unchanged. Consequently, induction of MazF toxin caused degradation of E.coli cellular mRNA but the recombinant gene transcription and protein synthesis continued, causing significant accumulation of high quality target protein. This expression system enables unparalleled signal to noise ratios that could dramatically simplify structural and functional studies of difficult-to-purify, biologically important proteins.
Tandem fusions and bacterial strain evolution for enhanced functional membrane protein production
Membrane protein production remains a significant challenge in its characterization and structure determination. Despite the fact that there are a variety of host cell types, E.coli remains the popular choice for producing recombinant membrane proteins. A group of scientists in Netherlands devised a robust strategy to increase the probability of functional membrane protein overexpression in E.coli.
By fusing Green Fluorescent Protein (GFP) and the Erythromycin Resistance protein (ErmC) to the C-terminus of a target membrane protein they wer e able to track the folding state of their target protein while using Erythromycin to select for increased expression. By increasing erythromycin concentration in the growth media and testing different membrane targets, they were able to identify four evolved E.coli strains, all of which carried a mutation in the hns gene, whose product is implicated in genome organization and transcriptional silencing. Through their experiments the group showed that partial removal of the transcriptional silencing mechanism was related to production of proteins that were essential for functional overexpression of membrane proteins.
The role of an anti-apoptotic factor in recombinant protein production
In a recent study, scientists at the Johns Hopkins University and Frederick National Laboratory for Cancer Research examined an alternative method of utilizing the benefits of anti-apoptotic gene expression to enhance the transient expression of biotherapeutics, specifically, through the co-transfection of Bcl-xL along with the product-coding target gene.
Chinese Hamster Ovary(CHO) cells were co-transfected with the product-coding gene and a vector containing Bcl-xL, using Polyethylenimine (PEI) reagent. They found that the cells co-transfected with Bcl-xL demonstrated reduced apoptosis, increased specific productivity, and an overall increase in product yield.
B-cell lymphoma-extra-large (Bcl-xL) is a mitochondrial transmembrane protein and a member of the Bcl-2 family of proteins which are known to act as either pro- or anti-apoptotic proteins. Bcl-xL itself acts as an anti-apoptotic molecule by preventing the release of mitochondrial contents such as cytochrome c, which would lead to caspase activation. Higher levels of Bcl-xL push a cell toward survival mode by making the membranes pores less permeable and leaky.
4.1 The Role and Importance of Transcription Factors
Larry H. Bernstein, MD, FCAP
4.2 RNA and the Transcription of the Genetic Code
Larry H. Bernstein, MD, FCAP
4.3 9:30 – 10:00, 6/13/2014, David Bartel “MicroRNAs, Poly(A) tails and Post-transcriptional Gene Regulation
Aviva Lev-Ari, PhD, RN
4.4 Transcriptional Silencing and Longevity Protein Sir2
Larry H. Bernstein, MD, FCAP
4.5 Ca2+ Signaling: Transcriptional Control
Larry H. Bernstein, MD, FCAP
4.6 Long Noncoding RNA Network regulates PTEN Transcription
Larry H. Bernstein, MD, FCAP
4.7 Zinc-Finger Nucleases (ZFNs) and Transcription Activator–Like Effector Nucleases (TALENs)
Larry H. Bernstein, MD, FCAP
4.8 Cardiac Ca2+ Signaling: Transcriptional Control
Larry H. Bernstein, MD, FCAP
4.9 Transcription Factor Lyl-1 Critical in Producing Early T-Cell Progenitors
Prabodh Kandala, PhD
4.10 Human Frontal Lobe Brain: Specific Transcriptional Networks
Aviva Lev-Ari, PhD, RN
4.11 Somatic, Germ-cell, and Whole Sequence DNA in Cell Lineage and Disease
Larry H. Bernstein, MD, FCAP
Summary of Transcription, Translation and Transcription Factors
Author and Curator: Larry H. Bernstein, MD, FCAP
Proteins are integral to the composition of the cytoskeleton, and also to the extracellular matrix. Many proteins are actually enzymes, carrying out the transformation of some substrate, a derivative of the food we ingest. They have a catalytic site, and they function with a cofactor – either a multivalent metal or a nucleotide. Proteins also are critically involved in the regulation of cell metabolism, and they are involved in translation of the DNA code, as they make up transcription factors (TFs). There are 20 essential amino acids that go into protein synthesis that are derived from animal or plant protein. Protein synthesis is carried out by the transport of mRNA out of the nucleus to the ribosome, where tRNA is paired with a matching amino acid, and the primary sequence of a protein is constructed as a linear string of amino acids.
This is illustrated in the following three pictures:
Proteins synthesized at distal locations frequently contain intrinsically disordered segments. These regions are generally rich in assembly-promoting modules and are often regulated by post-translational modifications. Such proteins are tightly regulated but display distinct temporal dynamics upon stimulation with growth factors. Thus, proteins synthesized on-site may rapidly alter proteome composition and act as dynamically regulated scaffolds to promote the formation of reversible cellular assemblies.
RJ Weatheritt, et al. Nature Structural & Molecular Biology 24 Aug, 2014; 21: 833–839 http://dx.do.orgi:/10.1038/nsmb.2876
Turquoise and red filled circle represents off-target and correct interaction partners, respectively. Wavy lines represent a disordered region within a distal site synthesis protein. Grey and red line in graphs represents profiles of t… http://www.nature.com/nsmb/journal/v21/n9/carousel/nsmb.2876-F5.jpg
In the the transcription process an RNA sequence is read. This is essential for protein synthesis through the ordering of the amino acids in the primary structure. However, there are microRNAs and noncoding RNAs, and there are transcription factors. The transcription factors bind to chromatin, and the RNAs also have some role in regulating the transcription process. (see picture above)
Transcription factors (TFs) interact dynamically in vivo with chromatin binding sites. Four different techniques are currently used to measure their kinetics in live cells,
- fluorescence recovery after photobleaching (FRAP),
- fluorescence correlation spectroscopy (FCS),
- single molecule tracking (SMT) and
- competition ChIP (CC).
A comparison of data from each of these techniques raises an important question:
- do measured transcription kinetics reflect biologically functional interactions at specific sites (i.e. working TFs) or
- do they reflect non-specific interactions (i.e. playing TFs)?
There are five key unresolved biological questions related to
- the functionality of transient and prolonged binding events at both
- specific promoter response elements as well as non-specific sites.
In support of functionality,
- there are data suggesting that TF residence times are tightly regulated, and
- that this regulation modulates transcriptional output at single genes.
In addition to this site-specific regulatory role, TF residence times
- also determine the fraction of promoter targets occupied within a cell
- thereby impacting the functional status of cellular gene networks.
- TF residence times, then, are key parameters that could influence transcription in multiple ways.
Quantifying transcription factor kinetics: At work or at play? Mueller F., et al. http://dx.doi.org:/10.3109/10409238.2013.833891
Dr. Virginie Mattot works in the team “Angiogenesis, endothelium activation and Cancer” directed by Dr. Fabrice Soncin at the Institut de Biologie de Lille in France where she studies the roles played by microRNAs in endothelial cells during physiological and pathological processes such as angiogenesis or endothelium activation. She has been using Target Site Blockers to investigate the role of microRNAs on putative targets.
A few years ago, the team identified
- an endothelial cell-specific gene which
- harbors a microRNA in its intronic sequence.
They have since been working on understanding the functions of
- both this new gene and its intronic microRNA in endothelial cells.
While they were searching for the functions of the intronic microRNA,
- theye identified an unknown gene as a putative target.
The aim of my project was to investigate if this unknown gene was actually a genuine target and
- if regulation of this gene by the microRNA was involved in endothelial cell function.
They had already shown the endothelial cell phenotype is associated with the inhibition of the intronic microRNA.
They then used miRCURY LNA™ Target Site Blockers to demonstrate
- the expression of this unknown gene is actually controlled by this microRNA.
- the microRNA regulates specific endothelial cell properties through regulation of this unknown gene.
MicroRNA function in endothelial cells – Solving the mystery of an unknown target gene using Target Site Blockers to investigate the role of microRNAs on putative targets
We first verified that this TSB was functional by analyzing
- the expression of the miRNA target against which the TSB was directed
- we then showed the TSB induced similar phenotypes as those when we inhibited the microRNA in the same cells.
Target Site Blockers were shown to be efficient tools to demonstrate the specific involvement of
- putative microRNA targets
- in the function played by this microRNA.
Some genes are known to have several different alternatively spliced protein variants, but the Scripps Research Institute’s Paul Schimmel and his colleagues have uncovered almost 250 protein splice variants of an essential, evolutionarily conserved family of human genes. The results were published July 17 in Science.
Focusing on the 20-gene family of aminoacyl tRNA synthetases (AARSs),
- the team captured AARS transcripts from human tissues—some fetal, some adult—and showed that
- many of these messenger RNAs (mRNAs) were translated into proteins.
Previous studies have identified several splice variants of these enzymes that have novel functions, but uncovering so many more variants was unexpected, Schimmel said. Most of these new protein products
- lack the catalytic domain but retain other AARS non-catalytic functional domains.
This study fundamentally effects how we view protein-synthesis, according to Michael Ibba (who was not involved in the work), The Scientist reported. “The unexpected and potentially vast expanded functional networks that emerge from this study have the potential to influence virtually any aspect of cell growth.”
The team—comprehensively captured and sequenced the AARS mRNAs from six human tissue types using high-throughput deep sequencing. They next showed that a proportion of these transcripts, including those missing the catalytic domain, indeed resulted in stable protein products:
- 48 of these splice variants associated with polysomes.
In vitro translation assays and the expression of more than 100 of these variants in cells confirmed that
- many of these variants could be made into stable protein products.
The AARS enzymes—of which there’s one for each of the 20 amino acids—bring together an amino acid with its appropriate transfer RNA (tRNA) molecule. This reaction allows a ribosome to add the amino acid to a growing peptide chain during protein translation. AARS enzymes can be found in all living organisms and are thought to be among the first proteins to have originated on Earth.
One goal of human genetics is to understand how the information for precise and dynamic gene expression programs is encoded in the genome. The interactions of transcription factors (TFs) with DNA regulatory elements clearly
- play an important role in determining gene expression outputs, yet
- the regulatory logic underlying functional transcription factor binding is poorly understood.
An important question in genomics is to understand how a class of proteins called ‘‘transcription factors’’ controls the expression level of other genes in the genome in a cell type-specific manner – a process that is essential to human development. One major approach to this problem is to study where these transcription factors bind in the genome, but this does not tell us about the effect of that binding on gene expression levels and
- it is generally accepted that much of the binding does not strongly influence gene expression.
DA Cusanovich et al. PLoS Genet 2014;10(3):e1004226. http://dx.doi.org:/10.1371/journal.pgen.1004226
We knocked down 59 TFs and chromatin modifiers in one HapMap lymphoblastoid cell line
- to evaluate the context of functional TF binding.
We then identified genes whose expression was affected by the knockdowns
- by intersecting the gene expression data with transcription factor binding data
(based on ChIP-seq and DNase-seq) - within 10 kb of the transcription start sites of expressed genes.
This combination of data allowed us to infer functional TF binding.
Only a small subset of genes bound by a factor were
- differentially expressed following the knockdown of that factor,
- suggesting that most interactions between TF and chromatin
- do not result in measurable changes in gene expression levels
- of putative target genes.
We found that functional TF binding is enriched
- in regulatory elements that harbor a large number of TF binding sites,
- at sites with predicted higher binding affinity, and
- at sites that are enriched in genomic regions annotated as ‘‘active enhancers.’’
We aim to be able to predict the expression pattern of a gene based on its regulatory
sequence alone.
Combining a TF knockdown approach with TF binding data can help us to
- distinguish functional binding from non-functional binding
This approach has previously been applied to the study of human TFs, although for the most part studies have only focused on
- the regulatory relationship of a single factor with its downstream targets.
The FANTOM consortium knocked down 52 different transcription factors in
- the THP-1 cell line, an acute monocytic leukemia-derived cell line, and
- used a subset of these to validate certain regulatory predictions based on binding motif enrichments.
We and others previously studied the regulatory architecture of gene expression in
- the model system of HapMap lymphoblastoid cell lines (LCLs) using both
- binding map strategies and QTL mapping strategies.
We now sought to use knockdown experiments targeting transcription factors in a HapMap LCL
- to refine our understanding of the gene regulatory circuitry of the human genome.
Therefore, We integrated the results of the knockdown experiments with previous data on TF binding to
- better characterize the regulatory targets of 59 different factors and
- to learn when a disruption in transcription factor binding
- is most likely to be associated with variation in the expression level of a nearby gene.
Gene expression levels following the knockdown were compared to
- expression data collected from six samples that were transfected with negative control siRNA.
Depending on the factor targeted, the knockdowns resulted in
- between 39 and 3,892 differentially expressed genes at an FDR of 5%
(Figure 1B; see Table S3 for a summary of the results).
The knockdown efficiency for the 59 factors ranged
- from 50% to 90% (based on qPCR; Table S1).
The qPCR measurements of the knockdown level were significantly
- correlated with estimates of the TF expression levels
- based on the microarray data (P =0.001; Figure 1C).
Did the factors tended to have a consistent effect (either up- or down-regulation)
- on the expression levels of genes they purportedly regulated?
All factors we tested are associated with both up- and down-regulation of downstream targets (Figure 6).
While there is compelling evidence for our inferences, the current chromatin functional annotations
- do not fully explain the regulatory effects of the knockdown experiments.
For example, the enrichments for binding in ‘‘strong enhancer’’ regions of the genome range from 7.2% to 50.1% (median = 19.2%),
- much beyond what is expected by chance alone, but far from accounting for all functional binding.
A slight majority of downstream target genes were expressed at higher levels
- following the knockdown for 15 of the 29 factors for which we had binding information (Figure 6B).
The factor that is associated with the largest fraction (68.8%) of up-regulated target genes following the knockdown is EZH2,
- the enzymatic component of the Polycomb group complex.
On the other end of the spectrum was JUND, a member of the AP-1 complex, for which
- 66.7% of differentially expressed targets were down-regulated following the knockdown.
Our results, combined with the previous work from our group and others make for a complicated view
- of the role of transcription factors in gene regulation as
- it seems difficult to reconcile the inference from previous work that
- many transcription factors should primarily act as activators with the results presented here.
One somewhat complicated hypothesis, which nevertheless can resolve the apparent discrepancy, is that
- the ‘‘repressive’’ effects we observe for known activators may be
- at sites in which the activator is acting as a weak enhancer of transcription and
- that reducing the cellular concentration of the factor
- releases the regulatory region to binding by an alternative, stronger activator.
Integrative study of Arabidopsis thaliana metabolomic and transcriptomic data
with the interactiveMarVis-Graph software
M Landesfeind, A Kaever, K Feussner, C Thurow, C Gatz, I Feussner and P Meinicke
PeerJ 2:e239; http://dx.doi.org /10.7717/peerj.239
High-throughput technologies notoriously generate large datasets often including data from different omics platforms. Each dataset contains data for several thousand experimental markers, e.g., mass-to-charge ratios in mass spectrometry or spots in DNA microarray analysis. An experimental marker is associated with an intensity profile which may include several measurements according to different experimental conditions (Dettmer, Aronov & Hammock, 2007).
The combined analysis and visualization of data from different high-throughput technologies remains a key challenge in bioinformatics.We present here theMarVis-Graph software for integrative analysis of metabolic and transcriptomic data. All experimental data is investigated in terms of the full metabolic network obtained from a reference database. The reactions of the network are scored based on the associated data, and
- sub-networks, according to connected high-scoring reactions, are identified.
Finally, MarVis-Graph scores the detected sub-networks,
- evaluates them by means of a random permutation test and
- presents them as a ranked list.
Furthermore, MarVis-Graph features an interactive network visualization that provides researchers with a convenient view on the results.
The key advantage ofMarVis-Graph is the analysis of reactions detached from their pathways so that
- it is possible to identify new pathways or
- to connect known pathways by previously unrelated reactions.
TheMarVis-Graph software is freely available for academic use and can be downloaded at: http://marvis.gobics.de/marvis-graph.
Significant differences or clusters may be explained by associated annotations, e.g., in terms of metabolic pathways or biological functions. During recent years, numerous specialized tools have been developed to aid biological researchers in automating all these steps (e.g., Medina et al., 2010; Kaever et al., 2009; Waegele et al., 2012). Comprehensive studies can be performed by combining technologies from different omics fields. The combination of transcriptomic and proteomic data sets revealed a strong
correlation between both kinds of data (Nie et al., 2007) and supported the detection of complex interactions, e.g., in RNA silencing (Haq et al., 2010). Moreover, correlations
were detected between RNA expression levels and metabolite abundances (Gibon et al., 2006). Therefore, tools that integrate, analyze and visualize experimental markers from different platforms are needed. To cope with the complexity of genome-wide studies, pathway models are utilized extensively as a simple abstraction of the underlying complex mechanisms. Set Enrichment Analysis (Subramanian et al., 2005) and Over-Representation Analysis (Huang, Sherman & Lempicki, 2009) have become state-of-the-art tools for analyzing large-scale datasets: both methods evaluate predefined sets of entities, e.g., the accumulation of differentially expressed genes in a pathway.
While manually curated pathways are convenient and easy to interpret, experimental studies have shown that all metabolic and signaling pathways are heavily interconnected (Kunkel & Brooks, 2002; Laule et al., 2003). Data from biomolecular databases support these studies: the metabolic network of Arabidopsis thaliana in the KEGG database (Kanehisa et al., 2012; Kanehisa & Goto, 2000) contains 1606 reactions from which 1464 are connected in a single sub-network (>91%), i.e., they
share a metabolite as product or substrate. In the AraCyc 10.0 database (Mueller, Zhang & Rhee, 2003; Rhee et al., 2006), more than 89% of the reactions are counted in a single sub-network. In both databases, most other reactions are completely disconnected. Additionally, Set Enrichment Analyses can not identify links between the predefined sets easily. This becomes even more important when analyzing smaller pathways as provided by the MetaCyc (Caspi et al., 2008; Caspi et al., 2012) database. Moreover, methods that utilize pathways as predefined sets ignore reactions and related biomolecular entities (e.g., metabolites, genes) which are not associated with a single pathway. For example, this affects 4000 reactions in MetaCyc and 2500 in KEGG, respectively (Altman et al., 2013). Therefore, it is desirable to develop additional methods
- that do not require predefined sets but may detect enriched sub-networks in the full metabolic network.
While several tools support the statistical analysis of experimental markers from one or more omics technologies and then utilize variants of Set Enrichment Analysis (Xia et al., 2012; Chen et al., 2013; Howe et al., 2011),
- no tool is able to explicitly search for connected reactions that include
- most of the metabolites, genes, and enyzmes with experimental evidence.
However, the automatic identification of sub-networks has been proven useful in other contexts, e.g., in the analysis of protein–protein-interaction networks (Alcaraz et al., 2012; Baumbach et al., 2012; Maeyer et al., 2013).
MarVis-Graph imports experimental markers from different high-throughput experiments and
- analyses them in the context of reaction-chains in full metabolic networks.
Then, MarVis-Graph scores the reactions in the metabolic network
- according to the number of associated experimental markers and
- identifies sub-networks consisting of subsequent, high-scoring reactions.
The resulting sub-networks are
- ranked according to a scoring method and visualized interactively.
Hereby, sub-networks consisting of reactions from different pathways may be identified to be important
- whereas the single pathways may not be found to be significantly enriched.
MarVis-Graph may also connect reactions without an assigned pathway
- to reactions within a particular pathway.
TheMarVis-Graph tool was applied in a case-study investigating the wound response in Arabidopsis thaliana to analyze combined metabolomic and transcriptomic high-throughput data.
Figure 1 Schema of the metabolic network representation in MarVis-Graph. Metabolite markers are shown in gray, metabolites in red, reactions in blue, enzymes in green, genes in yellow, transcript markers in pink, and pathways in turquoise color. The edges are shown in black with labels that comply with the biological meaning. The orange arrows depict the flow of score for the initial scoring (described in section “Initial Scoring”). (not shown)
In MarVis-Graph, metabolite markers obtained from mass-spectrometry experiments additionally contain the experimental mass. The experimental mass has to be
calculated based on the mass-to-charge ratio (m/z-value) and specific isotope- or adduct-corrections (Draper et al., 2009) by means of specialized tools, e.g.,MarVis-Filter
(Kaever et al., 2012).
For each transcript marker the corresponding annotation has to be given. In DNA microarray experiments, each spot (transcript marker) is specific for a gene and can
therefore be used for annotation. For other technologies an annotation has to be provided by external tools.
In MarVis-Graph, each reaction is scored initially based on the associated experimental data (see “Initial scoring”). This initial scoring is refined (see “Refining the scoring”) and afterwards reactions with a score below a user-defined threshold are removed. The network is
- decomposed into subsequent high-scoring reactions that constitute the sub-networks.
The weight of each experimental marker (see “Experimental markers”) is equally distributed over all metabolites and genes associated with the metabolite marker or
transcript marker, respectively. For all vertices, this is repeated as illustrated in Fig. 1 until the weights are accumulated by the reactions.
The initial reaction scores are used as input scoring for the random walk algorithm. The algorithm is performed as described by Glaab et al. (2012) with a user-defined
restart-probability r (default value 0.8). After convergence of the algorithm, reactions with a score lower than the user-defined threshold t (default value t = 1−r) are removed from the reaction network. During the removal process,
- the network is decomposed into pairwise disconnected sub-networks containing only high-scoring reactions.
In the following, a resulting sub-network is denoted by a prime: G′ = (V′,L′) with V′ = M′ ∪C′ ∪R′ ∪E′ ∪G′ ∪T′ ∪P′.
The scores of the identified sub-networks can be assessed using a random permutation test, evaluating the marker annotations under the null hypothesis of being connected
randomly. Here, the assignments
- from metabolite markers to metabolites and from transcript markers to genes are randomized.
For each association between a metabolite marker and a metabolite,
- this connection is replaced by a connection between a randomly chosen metabolite marker and a randomly chosen metabolite.
The random metabolite marker is chosen from the pool of formerly connected metabolite markers. Each connected transcript marker
- is associated with a randomly chosen gene.
Choosing from the list of already connected experimental markers ensures that
- the sum of weights from the original and the permuted network are equal.
This method differs from the commonly utilized XSwap permutation (Hanhij¨arvi, Garriga & Puolam¨aki, 2009) that is based on swapping endpoints of two random edges. The main difference of our permutation method is that it results in a network with different topological structure, i.e., different degree of the metabolite and gene nodes.
Finally, the sub-networks are detected and scored with the same parameters applied for the original network. Based on the scores of the networks identified in the random
permutations, the family-wise-error-rate (FWER) and false-discovery-rate (FDR) are calculated for each originally identified sub-network.
MarVis-Graph was applied in a case study investigating the A. thaliana wound response. Data from a metabolite fingerprinting (Meinicke et al., 2008) and a DNA microarray
experiment (Yan et al., 2007) were imported into a metabolic network specific for A. thaliana created from the AraCyc 10.0 database (Lamesch et al., 2011). The metabolome
and transcriptome have been measured before wounding as control and at specific time points after wounding in wild-type and in the allene oxide synthase (AOS) knock-out
mutant dde-2-2 (Park et al., 2002) of A. thaliana Columbia (see Table 1). The AOS mutant was chosen, because AOS catalyzes the first specific step in the biosynthesis of the hormone jasmonic acid, which is the key regulator in wound response of plants (Wasternack & Hause, 2013).
Both datasets have been preprocessed with theMarVis-Filter tool (Kaever et al., 2012) utilizing the Kruskal–Wallis p-value calculation on the intensity profiles. Based on the ranking of ascending p-values,
- the first 25% of the metabolite markers and 10% of the transcript markers have been selected for further investigation (Data S2).
The filtered metabolite and transcript markers were imported into the metabolic network. For metabolite markers, metabolites were associated
- if the metabolite marker’s detected mass differs from the metabolites monoisotopic mass by a maximum of 0.005u.
Transcript markers were linked to the genes whose ID equaled the ID given in the CATMA database (Sclep et al., 2007) for that transcript marker.
Table 2 Vertices in the A. thaliana specific metabolic network after import of experimental markers. Number of objects in the metabolic network
in absolute counts and relative abundances. For experimental markers, the with annotation column gives the number of metabolite markers and
transcript markers that were annotated with a metabolite or gene, respectively. The direct evidence column contains the number of metabolites
and genes, that are associated with a metabolite marker or transcript marker. For enzymes, this is the number of enzymes encoded by a gene with
direct evidence. The number of vertices with an association to a reaction is given in the with reaction column. In the last column, this is given for
associations to metabolic pathways. (not shown)
MarVis-Graph detected a total of 133 sub-networks. The sub-networks were ranked according to size Ss, diameter Sd, and sum-of-weights Ssow
scores (Table S4). Interestingly, the different rankings show a high correlation with all pairwise correlations higher than 0.75 (Pearson correlation
coefficient) and 0.6 (Spearman rank correlation).
Allene-oxide cyclase sub-network
In all rankings, the sub-network allene-oxide cyclase (named after the reaction with the highest score in this sub-network) appeared as top candidate.
This sub-network is constituted of reactions from different pathways related to fatty acids. Figure 2 shows a visualization of the sub-network.
Jasmonic acid biosynthesis. The main part of the sub-network is formed by reactions from the “jasmonic acid biosynthesis” (PlantMetabolic Network, 2013)
resulting in jasmonic acid (jasmonate). The presence of this pathway is very well established because of its central role in mediating the plants wound response
(Reymond & Farmer, 1998; Creelman, Tierney & Mullet, 1992). Additionally, metabolites and transcripts from this pathway were expected to show prominent
expression profiles because AOS, a key enzyme in this pathway, is knocked-out in themutant plant. Jasmonic acid derivatives and hormones.
Jasmonic acid derivatives and hormones. Jasmonate is a precursor for a broad variety of plant hormones (Wasternack & Hause, 2013), e.g., the derivative (-)-
jasmonic acid methyl ester (also Methyl Jasmonic Acid; MeJA) is a volatile, airborne signal mediating wound response between plants (Farmer&Ryan, 1990).
Reactions from the jasmonoyl-amino acid conjugates biosynthesis I (PMN, 2013a) pathway connect jasmonate to different amino acids, including L-valine,
L-leucine, and L-isoleucine. Via these amino acids, this sub-network is connected to the indole-3-acetylamino acid biosynthesis (PMN, 2013b) (IAA biosynthesis).
Again, this pathway produces a well known plant hormone: Auxine (Woodward & Bartel, 2005). Even though, jasmonate and auxin are both plant hormones, their
connection in this subnetwork is of minor relevance because amino acid conjugates are often utilized as active or storage forms of signaling molecules.While
jasmonoyl-amino acid conjugates represent the active signaling form of jasmonates, IAA amino acid conjugates are the storage form of this hormone (Staswick et al.,
2005).

Figure 2 Schema of the allene-oxide cyclase sub-network. Metabolites are shown in red, reactions in blue, and enzymes in green color. Metabolites and reactions without direct experimental evidence are marked by a dashed outline and a brighter color while enzymes without experimental evidence are hidden. The metabolic pathways described in section “Resulting sub-networks” are highlighted with different colors. The orange and green parts indicate the reaction chains required to build jasmonate and its amino acid conjugates. The coloring of pathways was done manually after export from MarVis-Graph.
The ω-3-fatty acid desaturase should catalyze a reaction from linoleate to α-linolenate. Metabolite markers that match the mass of crepenynic acid do also match α-linolenate
because both molecules have the same sum-formula and monoisotopic mass. As mentioned above, MarVis-Graph compiled the metabolic network for this study
from the AraCyc database version 10.0. On June 4th, a curator changed the database to remove theΔ12-fatty acid dehydrogenase prior to the release of AraCyc version 11.0.
The presented new software tool MarVis-Graph supports the investigation and visualization of omics data from different fields of study. The introduced algorithm for
identification of sub-networks is able to identify reaction-chains across different pathways and includes reactions that are not associated with a single pathway. The application of MarVis-Graph in the case study on A. thaliana wound response resulted in a convenient graphical representation of high-throughput data which allows the analysis of the complex dynamics in a metabolic network.
Chapter 5: Sub-cellular Structure
Introduction to Subcellular Structure
Author and Curator: Larry H. Bernstein, MD, FCAP
The following chapter of the metabolism/transcriptomics/proteomics/metabolomics series deals with the subcellular structure of the cell. This would have to include the cytoskeleton, which has a key role in substrate and ion efflux and influx, and in cell movement mediated by tubulins. It has been extensively covered already. Much of the contributions here are concerned with the mitochondrion, which is also covered in metabolic pathways. The ribosome is the organelle that we have discussed with respect to the transcription and translation of the genetic code through mRNA and tRNA, and the therapeutic implications of SiRNA as well as the chromatin regulation of lncRNA.
We have also encountered the mitochondrion and the lysosome in the discussion of apoptosis and autophagy, maintaining the balance between cell regeneration and cell death.
I here list the organelles:
- Nucleus
- Centrosome
- Nuclear Membrane
- Ribososome
- Endoplasmic Reticulum
- Mitochondria
- Lysosome
- Cytoskeleton
- Golgi apparatus
- Cytoplasm

http://www.youtube.com/watch?feature=player_embedded&v=JufLDxmCwB0
http://www.youtube.com/watch?feature=player_embedded&v=FFrKN7hJm64
Golgi Apparatus
Found within the cytoplasm of both plant and animal cells, the Golgi is composed of stacks of membrane-bound structures known as cisternae (singular: cisterna). An individual stack is sometimes called a dictyosome (from Greek dictyon: net + soma: body), especially in plant cells. A mammalian cell typically contains 40 to 100 stacks. Between four and eight cisternae are usually present in a stack; however, in some protists as many as sixty have been observed. Each cisterna comprises a flat, membrane-enclosed disc that includes special Golgi enzymes which modify or help to modify cargo proteins that travel through it.
The cisternae stack has four functional regions: the cis-Golgi network, medial-Golgi, endo-Golgi, and trans-Golgi network. Vesicles from the endoplasmic reticulum (via the vesicular-tubular clusters) fuse with the network and subsequently progress through the stack to the trans-Golgi network, where they are packaged and sent to their destination.
The Golgi apparatus is integral in modifying, sorting, and packaging these macromolecules for cell secretion (exocytosis) or use within the cell. It primarily modifies proteins delivered from the rough endoplasmic reticulum, but is also involved in the transport of lipids around the cell, and the creation of lysosomes. Enzymes within the cisternae are able to modify the proteins by addition of carbohydrates (glycosylation) and phosphates (phosphorylation). In order to do so, the Golgi imports substances such as nucleotide sugars from the cytosol. These modifications may also form a signal sequence which determines the final destination of the protein. For example, the Golgi apparatus adds a mannose-6-phosphate label to proteins destined for lysosomes.
The Golgi plays an important role in the synthesis of proteoglycans, which are molecules present in the extracellular matrix of animals. It is also a major site ofcarbohydrate synthesis. This includes the production of glycosaminoglycans (GAGs), long unbranched polysaccharides which the Golgi then attaches to a protein synthesised in the endoplasmic reticulum to form proteoglycans. Enzymes in the Golgi polymerize several of these GAGs via a xylose link onto the core protein. Another task of the Golgi involves the sulfation of certain molecules passing through its lumen via sulfotranferases that gain their sulfur molecule from a donor called PAPS. This process occurs on the GAGs of proteoglycans as well as on the core protein. Sulfation is generally performed in the trans-Golgi network. The level of sulfation is very important to the proteoglycans’ signalling abilities, as well as giving the proteoglycan its overall negative charge.
The phosphorylation of molecules requires that ATP is imported into the lumen of the Golgi and utilised by resident kinases such as casein kinase 1 and casein kinase 2. One molecule that is phosphorylated in the Golgi is apolipoprotein, which forms a molecule known as VLDL that is found in plasma. It is thought that the phosphorylation of these molecules labels them for secretion into the blood.
The Golgi has a putative role in apoptosis, with several Bcl-2 family members localised there, as well as to the mitochondria. A newly characterized protein, GAAP (Golgi anti-apoptotic protein), almost exclusively resides in the Golgi and protects cells from apoptosis by an as-yet undefined mechanism.
The vesicles that leave the rough endoplasmic reticulum are transported to the cisface of the Golgi apparatus, where they fuse with the Golgi membrane and empty their contents into the lumen. Once inside the lumen, the molecules are modified, then sorted for transport to their next destinations. The Golgi apparatus tends to be larger and more numerous in cells that synthesize and secrete large amounts of substances; for example, the plasma B cells and the antibody-secreting cells of the immune system have prominent Golgi complexes.
Those proteins destined for areas of the cell other than either the endoplasmic reticulum or Golgi apparatus are moved towards the trans face, to a complex network of membranes and associated vesicles known as the trans-Golgi network(TGN). This area of the Golgi is the point at which proteins are sorted and shipped to their intended destinations by their placement into one of at least three different types of vesicles, depending upon the molecular marker they carry.

Diagram of secretory process from endoplasmic reticulum (orange) to Golgi apparatus (pink). 1. Nuclear membrane; 2. Nuclear pore; 3. Rough endoplasmic reticulum (RER); 4. Smooth endoplasmic reticulum (SER); 5. Ribosome attached to RER; 6. Macromolecules; 7. Transport vesicles; 8. Golgi apparatus; 9. Cis face of Golgi apparatus; 10. Trans face of Golgi apparatus; 11. Cisternae of the Golgi Apparatus
Exocytotic vesicles
After packaging, the vesicles bud off and immediately move towards the plasma membrane, where they fuse and release the contents into the extracellular space in a process known as constitutive secretion. (Antibody release by activated plasma B cells)
Secretory vesicles
After packaging, the vesicles bud off and are stored in the cell until a signal is given for their release. When the appropriate signal is received they move towards the membrane and fuse to release their contents. This process is known as regulated secretion. (Neurotransmitter release from neurons)
Lysosomal vesicles
Vesicle contains proteins and ribosomes destined for the lysosome, an organelle of degradation containing many acid hydrolases, or to lysosome-like storage organelles. These proteins include both digestive enzymes and membrane proteins. The vesicle first fuses with the late endosome, and the contents are then transferred to the lysosome via unknown mechanisms.
http://en.wikipedia.org/wiki/Golgi_apparatus
Lysosome (derived from the Greek words lysis, meaning “to loosen”, and soma, “body”) is a membrane-bound cell organelle found in animal cells (they are absent inred blood cells). They are structurally and chemically spherical vesicles containinghydrolytic enzymes, which are capable of breaking down virtually all kinds ofbiomolecules, including proteins, nucleic acids, carbohydrates, lipids, and cellular debris. Lysosomes are responsible for cellular homeostasis for their involvements in secretion, plasma membrane repair, cell signalling and energy metabolism, which are related to health and diseases. Depending on their functional activity their sizes can be very different, as the biggest ones can be more than 10 times bigger than the smallest ones. They were discovered and named by Belgian biologist Christian de Duve, who eventually received the Nobel Prize in Physiology or Medicine in 1974.
Enzymes of the lysosomes are synthesised in the rough endoplasmic reticulum. The enzymes are released from Golgi apparatus in small vesicles which ultimately fuse with acidic vesicles called endosomes, thus becoming full lysosomes. In the process the enzymes are specifically tagged with mannose 6-phosphate to differentiate them from other enzymes. Lysosomes are interlinked with three intracellular processes namely phagocytosis, endocytosis and autophagy. Extracellular materials such asmicroorganisms taken up by phagocytosis, macromolecules by endocytosis, and unwanted cell organelles are fused with lysosomes in which they are broken down to their basic molecules. Thus lysosomes are the recycling units of a cell.
http://en.wikipedia.org/wiki/Lysosome
The endoplasmic reticulum (ER) is a type of organelle in the cells of eukaryotic organisms that forms an interconnected network of flattened, membrane-enclosed sacs or tubes known as cisternae. The membranes of the ER are continuous with the outer membrane of the nuclear envelope. Endoplasmic reticulum occurs in most types of eukaryotic cells, including the most primitive Giardia, but is absent from red blood cells and spermatozoa. There are two types of endoplasmic reticulum, rough endoplasmic reticulum (RER) and smooth endoplasmic reticulum (SER). The outer (cytosolic) face of the rough endoplasmic reticulum is studded with ribosomes that are the sites of protein synthesis. The rough endoplasmic reticulum is especially prominent in cells such as hepatocytes where active smooth endoplasmic reticulum lacks ribosomes and functions in lipid metabolism, carbohydrate metabolism, anddetoxification and is especially abundant in mammalian liver and gonad cells. The lacey membranes of the endoplasmic reticulum were first seen in 1945 by Keith R. Porter, Albert Claude, Brody Meskers and Ernest F. Fullam, using electron microscopy.
http://en.wikipedia.org/wiki/Endoplasmic_reticulum

https://2cslacardano.wikispaces.com/file/view/Cell7.png/338811858/408×313/Cell7.png
{kind=link}
Cytoskeleton
The Effects of Actomyosin Tension on Nuclear Pore Transport
Rachel Sammons
Undergraduate Honors Thesis
Spring 2011
The cytoskeleton maintains cellular structure and tension through a force balance with the nucleus, where actomyosin is anchored to the nuclear envelope by nesprin integral proteins. It is hypothesized that the presence or absence of this tension alters the transport of molecules through the nuclear pore complex. We tested the effects of cytoskeletal tension on nuclear transport in human umbilical vein endothelial cells (HUVECs) by performing fluorescence recovery after photo-bleaching (FRAP) experiments on the nuclei to monitor the passive transport of the molecules through nuclear pores.
Using myosin inhibitors, as well as siRNA transfections to reduce the expression of nesprin-1, we altered the nucleo-cytoskeletal force balance and monitored the effect of each on the nuclear pore. FRAP data was fit to a diffusion model by assuming pseudo-steady state inside the nuclear pore, perfect mixing within both the cytoplasm and the nucleus, and no intracellular binding of the fluorescent probes. From these results and a model from the current literature relating diffusion rate constants to nuclear pore radii, we were able to determine that changing cytoskeletal tension alters nuclear pore size and passive transport.

image of nuclear pores on the external surface of the nuclear envelope

nuclear envelope and FG filaments
Figure 1: The structure and location of the nuclear pore, shown by (a) AFM image of nuclear pores on the external surface of the nuclear envelope[5] and (b) computer model cross-section. The nuclear envelope is shown in cyan, and FG filaments in blue can be seen throughout the channel. The nuclear basket extends into the nucleoplasm.
Fusion-pore expansion during syncytium formation is restricted by an actin network
A Chen, E Leikina, K Melikov, B Podbilewicz, MM. Kozlov and LV. Chernomordik,*
J Cell Sci 1 Nov 2008;121: 3619-3628. http://dx.doi.org:/10.1242/jcs.032169
Effects of actin-modifying agents indicate that the actin cortex slows down pore expansion. We propose that the growth of the strongly bent fusion-pore rim is restricted by a dynamic resistance of the actin network and driven by membrane-bending proteins that are involved in the generation of highly curved intracellular membrane compartments.
5.1 Mitochondria: Origin from Oxygen free environment, Role in Aerobic Glycolysis and Metabolic Adaptation
Larry H Bernstein, MD, FCAP
5.2 Mitochondrial Metabolism and Cardiac Function
Larry H. Bernstein, MD, FCAP
5.3 Mitochondria: More than just the “Powerhouse of the Cell”
Ritu Saxena, PhD
5.4 Mitochondrial Fission and Fusion: Potential Therapeutic Targets?
Ritu Saxena, PhD
5.5 Mitochondrial Mutation Analysis might be “1-step” Away
Ritu Saxena, PhD
5.6 Autophagy-Modulating Proteins and Small Molecules Candidate Targets for Cancer Therapy: Commentary of Bioinformatics Approaches
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
5.7 Chromatophagy, A New Cancer Therapy: Starve The Diseased Cell Until It Eats Its Own DNA
Aviva Lev-Ari, PhD, RN
5.8 A Curated Census of Autophagy-Modulating Proteins and Small Molecules Candidate Targets for Cancer Therapy
Aviva Lev-Ari, PhD, RN
5.9 Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility
Larry H Bernstein, MD, FCAP, Stephen Williams, PhD, and Aviva Lev-Ari, PhD, RN
Summary of Cell Structure, Anatomic Correlates of Metabolic Function
Author and Curator: Larry H. Bernstein, MD, FCAP
This chapter has been concerned with the subcellular ultrastructure of organelles, and importantly, their function. There is no waste in the cell structure. The nucleus has the instructions necessary to carry out the cell’s functions. In the Eukaryotic cell there is significant differentiation so that the cells are regulated for the needs that they uniquely carry out. When there is disregulation, it leads to remodeling or to cell death.
Here I shall note some highlights of this chapter.
- In every aspect of cell function, proteins are involved embedded in the structure, for most efficient functioning.
- Metabolic regulation is dependent on pathways that are also linkages of proteins.
- Energy utilization is dependent on enzymatic reactions, often involving essential metal ions of high valence numbers, which facilitates covalent and anion binding, and has an essential role in allostericity.
Mitochondria

http://en.wikipedia.org/wiki/File:Mitochondria,_mammalian_lung_-_TEM.jpg
{kind=link}
Mitochondria range from 0.5 to 1.0 micrometer (μm) in diameter. These structures are sometimes described as “cellular power plants” because they generate most of the cell’s supply of adenosine triphosphate (ATP), used as a source of chemical energy. In addition to supplying cellular energy, mitochondria are involved in other tasks such as signaling, cellular differentiation, cell death, as well as the control of the cell cycle and cell growth. Mitochondria have been implicated in several human diseases, including mitochondrial disorders and cardiac dysfunction.
The number of mitochondria in a cell can vary widely by organism, tissue, and cell type. For instance, red blood cells have no mitochondria, whereas liver cells can have more than 2000. The organelle is composed of compartments that carry out specialized functions. These compartments or regions include the outer membrane, the intermembrane space, the inner membrane, and the cristae and matrix. Mitochondrial proteins vary depending on the tissue and the species. The mitochondrial proteome is thought to be dynamically regulated. Although most of a cell’s DNA is contained in the cell nucleus, the mitochondrion has its own independent genome. Further, its DNA shows substantial similarity to bacterialgenomes.
In 1913 particles from extracts of guinea-pig liver were linked to respiration by Otto Heinrich Warburg, which he called “grana”. Warburg and Heinrich Otto Wieland, who had also postulated a similar particle mechanism, disagreed on the chemical nature of the respiration. It was not until 1925 when David Keilin discovered cytochromesthat the respiratory chain was described. In 1939, experiments using minced muscle cells demonstrated that one oxygen atom can form two adenosine triphosphate molecules, and, in 1941, the concept of phosphate bonds being a form of energy in cellular metabolism was developed by Fritz Albert Lipmann. In the following years, the mechanism behind cellular respiration was further elaborated, although its link to the mitochondria was not known. The introduction of tissue fractionation by Albert Claude allowed mitochondria to be isolated from other cell fractions and biochemical analysis to be conducted on them alone. In 1946, he concluded that cytochrome oxidase and other enzymes responsible for the respiratory chain were isolated to the mitchondria.
The first high-resolution micrographs appeared in 1952, replacing the Janus Green stains as the preferred way of visualising the mitochondria. This led to a more detailed analysis of the structure of the mitochondria, including confirmation that they were surrounded by a membrane. It also showed a second membrane inside the mitochondria that folded up in ridges dividing up the inner chamber and that the size and shape of the mitochondria varied from cell to cell. In 1967, it was discovered that mitochondria contained ribosomes. In 1968, methods were developed for mapping the mitochondrial genes, with the genetic and physical map of yeast mitochondria being completed in 1976.
A mitochondrion contains outer and inner membranes composed of phospholipid bilayers and proteins. The two membranes have different properties. Because of this double-membraned organization, there are five distinct parts to a mitochondrion. They are:
- the outer mitochondrial membrane,
- the intermembrane space (the space between the outer and inner membranes),
- the inner mitochondrial membrane,
- the cristae space (formed by infoldings of the inner membrane), and
- the matrix (space within the inner membrane).
Mitochondria stripped of their outer membrane are called mitoplasts.

{kind=link}
Mitochondrion ultrastructure (interactive diagram) A mitochondrion has a double membrane; the inner one contains its chemiosmotic apparatus and has deep grooves which increase its surface area. While commonly depicted as an “orange sausage with a blob inside of it” (like it is here), mitochondria can take many shapes and their intermembrane space is quite thin.
The intermembrane space is the space between the outer membrane and the inner membrane. It is also known as perimitochondrial space. Because the outer membrane is freely permeable to small molecules, the concentrations of small molecules such as ions and sugars in the intermembrane space is the same as thecytosol. However, large proteins must have a specific signaling sequence to be transported across the outer membrane, so the protein composition of this space is different from the protein composition of the cytosol. One protein that is localized to the intermembrane space in this way is cytochrome c.
The inner mitochondrial membrane contains proteins with five types of functions:
- Those that perform the redox reactions of oxidative phosphorylation
- ATP synthase, which generates ATP in the matrix
- Specific transport proteins that regulate metabolite passage into and out of the matrix
- Protein import machinery.
- Mitochondria fusion and fission protein.
It contains more than 151 different polypeptides, and has a very high protein-to-phospholipid ratio (more than 3:1 by weight, which is about 1 protein for 15 phospholipids). The inner membrane is home to around 1/5 of the total protein in a mitochondrion. In addition, the inner membrane is rich in an unusual phospholipid,cardiolipin. This phospholipid was originally discovered in cow hearts in 1942, and is usually characteristic of mitochondrial and bacterial plasma membranes. Cardiolipin contains four fatty acids rather than two, and may help to make the inner membrane impermeable. Unlike the outer membrane, the inner membrane doesn’t contain porins, and is highly impermeable to all molecules. Almost all ions and molecules require special membrane transporters to enter or exit the matrix. Proteins are ferried into the matrix via the translocase of the inner membrane (TIM) complex or via Oxa1. In addition, there is a membrane potential across the inner membrane, formed by the action of the enzymes of the electron transport chain.
The inner mitochondrial membrane is compartmentalized into numerous cristae, which expand the surface area of the inner mitochondrial membrane, enhancing its ability to produce ATP. For typical liver mitochondria, the area of the inner membrane is about five times as large as the outer membrane. This ratio is variable and mitochondria from cells that have a greater demand for ATP, such as muscle cells, contain even more cristae. These folds are studded with small round bodies known as F1 particles or oxysomes. These are not simple random folds but rather invaginations of the inner membrane, which can affect overall chemiosmoticfunction. One recent mathematical modeling study has suggested that the optical properties of the cristae in filamentous mitochondria may affect the generation and propagation of light within the tissue.

{kind=link}
The matrix is the space enclosed by the inner membrane. It contains about 2/3 of the total protein in a mitochondrion. The matrix is important in thThe MAM is enriched in enzymes involved in lipid biosynthesis, such as phosphatidylserine synthase on the ER face and phosphatidylserine decarboxylase on the mitochondrial face.[28][29] Because mitochondria are dynamic organelles constantly undergoingfission and fusion events, they require a constant and well-regulated supply of phospholipids for membrane integrity.[30][31] But mitochondria are not only a destination for the phospholipids they finish synthesis of; rather, this organelle also plays a role in inter-organelle trafficking of the intermediates and products of phospholipid biosynthetic pathways, ceramide and cholesterol metabolism, and glycosphingolipid anabolisme production of ATP with the aid of the ATP synthase contained in the inner membrane. The matrix contains a highly concentrated mixture of hundreds of enzymes, special mitochondrial ribosomes, tRNA, and several copies of the mitochondrial DNA genome. Of the enzymes, the major functions include oxidation of pyruvate and fatty acids, and the citric acid cycle.
Purified MAM from subcellular fractionation has shown to be enriched in enzymes involved in phospholipid exchange, in addition to channels associated with Ca2+signaling. The mitochondria-associated ER membrane (MAM) is another structural element that is increasingly recognized for its critical role in cellular physiology andhomeostasis. Once considered a technical snag in cell fractionation techniques, the alleged ER vesicle contaminants that invariably appeared in the mitochondrial fraction have been re-identified as membranous structures derived from the MAM—the interface between mitochondria and the ER. Physical coupling between these two organelles had previously been observed in electron micrographs and has more recently been probed with fluorescence microscopy. Such studies estimate that at the MAM, which may comprise up to 20% of the mitochondrial outer membrane, the ER and mitochondria are separated by a mere 10–25 nm and held together by protein tethering complexes.
Such trafficking capacity depends on the MAM, which has been shown to facilitate transfer of lipid intermediates between organelles. In contrast to the standard vesicular mechanism of lipid transfer, evidence indicates that the physical proximity of the ER and mitochondrial membranes at the MAM allows for lipid flipping between opposed bilayers. Despite this unusual and seemingly energetically unfavorable mechanism, such transport does not require ATP. Instead, in yeast, it has been shown to be dependent on a multiprotein tethering structure termed the ER-mitochondria encounter structure, or ERMES, although it remains unclear whether this structure directly mediates lipid transfer or is required to keep the membranes in sufficiently close proximity to lower the energy barrier for lipid flipping.
A critical role for the ER in calcium signaling was acknowledged before such a role for the mitochondria was widely accepted, in part because the low affinity of Ca2+channels localized to the outer mitochondrial membrane seemed to fly in the face of this organelle’s purported responsiveness to changes in intracellular Ca2+ flux. But the presence of the MAM resolves this apparent contradiction: the close physical association between the two organelles results in Ca2+ microdomains at contact points that facilitate efficient Ca2+ transmission from the ER to the mitochondria. Transmission occurs in response to so-called “Ca2+ puffs” generated by spontaneous clustering and activation of IP3R, a canonical ER membrane Ca2+channel.
The properties of the Ca2+ pump SERCA and the channel IP3R present on the ER membrane facilitate feedback regulation coordinated by MAM function. In particular, clearance of Ca2+ by the MAM allows for spatio-temporal patterning of Ca2+signaling because Ca2+ alters IP3R activity in a biphasic manner. SERCA is likewise affected by mitochondrial feedback: uptake of Ca2+ by the MAM stimulates ATP production, thus providing energy that enables SERCA to reload the ER with Ca2+for continued Ca2+ efflux at the MAM. Thus, the MAM is not a passive buffer for Ca2+ puffs; rather it helps modulate further Ca2+ signaling through feedback loops that affect ER dynamics.
Regulating ER release of Ca2+ at the MAM is especially critical because only a certain window of Ca2+ uptake sustains the mitochondria, and consequently the cell, at homeostasis. Sufficient intraorganelle Ca2+ signaling is required to stimulate metabolism by activating dehydrogenase enzymes critical to flux through the citric acid cycle. However, once Ca2+ signaling in the mitochondria passes a certain threshold, it stimulates the intrinsic pathway of apoptosis in part by collapsing the mitochondrial membrane potential required for metabolism. Studies examining the role of pro- and anti-apoptotic factors support this model; for example, the anti-apoptotic factor Bcl-2 has been shown to interact with IP3Rs to reduce Ca2+ filling of the ER, leading to reduced efflux at the MAM and preventing collapse of the mitochondrial membrane potential post-apoptotic stimuli. Given the need for such fine regulation of Ca2+ signaling, it is perhaps unsurprising that dysregulated mitochondrial Ca2+ has been implicated in several neurodegenerative diseases, while the catalogue of tumor suppressors includes a few that are enriched at the MAM.
…more
http://en.wikipedia.org/wiki/Mitochondrion
Lysosome and Apoptosis
Role of autophagy in cancer
R Mathew, V Karantza-Wadsworth & E White
Nature Reviews Cancer 7, 961-967 (Dec 2007) | http://dx.doi.org:/10.1038/nrc2254
Autophagy is a cellular degradation pathway for the clearance of damaged or superfluous proteins and organelles. The recycling of these intracellular constituents also serves as an alternative energy source during periods of metabolic stress to maintain homeostasis and viability. In tumour cells with defects in apoptosis, autophagy allows prolonged survival. Paradoxically, autophagy defects are associated with increased tumorigenesis, but the mechanism behind this has not been determined. Recent evidence suggests that autophagy provides a protective function to limit tumour necrosis and inflammation, and to mitigate genome damage in tumour cells in response to metabolic stress.
Sustained Activation of mTORC1 in Skeletal Muscle Inhibits Constitutive and Starvation-Induced Autophagy and Causes a Severe, Late-Onset Myopathy
P Castets, S Lin, N Rion, S Di Fulvio, et al.
cell-metabolism 7 May, 2013; 17(5): p731–744 http://dx.doi.org/10.1016/j.cmet.2013.03.015
- mTORC1 inhibition is required for constitutive and starvation-induced autophagy
- Sustained activation of mTORC1 causes a severe myopathy due to autophagy impairment
- TSC1 depletion is sufficient to activate mTORC1 irrespective of other stimuli
- mTORC1 inactivation is sufficient to trigger LC3 lipidation
Autophagy is a catabolic process that ensures homeostatic cell clearance and is deregulated in a growing number of myopathological conditions. Although FoxO3 was shown to promote the expression of autophagy-related genes in skeletal muscle, the mechanisms triggering autophagy are unclear. We show that TSC1-deficient mice (TSCmKO), characterized by sustained activation of mTORC1, develop a late-onset myopathy related to impaired autophagy. In young TSCmKO mice,
- constitutive and starvation-induced autophagy is blocked at the induction steps via
- mTORC1-mediated inhibition of Ulk1, despite FoxO3 activation.
Rapamycin is sufficient to restore autophagy in TSCmKO mice and
- improves the muscle phenotype of old mutant mice.
Inversely, abrogation of mTORC1 signaling by
- depletion of raptor induces autophagy regardless of FoxO inhibition.
Thus, mTORC1 is the dominant regulator of autophagy induction in skeletal muscle and
- ensures a tight coordination of metabolic pathways.
These findings may open interesting avenues for therapeutic strategies directed toward autophagy-related muscle diseases.
Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice
Viviana Moresi, et al. PNAS Jan 31, 2012; 109(5): 1649-1654
http://dx.doi.org:/10.1073/pnas.1121159109
http://www.pnas.org/content/109/5/1649/F6.medium.gif
{kind=link}
HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy
Beharry, PB. Sandesara, BM. Roberts, et al.
J. Cell Sci. Apr 2014 127 (7) 1441-1453 http://dx.doi.org:/10.1242/jcs.136390
The Forkhead box O (FoxO) transcription factors are activated, and necessary for the muscle atrophy, in several pathophysiological conditions, including muscle disuse and cancer cachexia. However, the mechanisms that lead to FoxO activation are not well defined. Recent data from our laboratory and others indicate that
- the activity of FoxO is repressed under basal conditions via reversible lysine acetylation,
- which becomes compromised during catabolic conditions.
Therefore, we aimed to determine how histone deacetylase (HDAC) proteins contribute to
- activation of FoxO and induction of the muscle atrophy program.
Through the use of various pharmacological inhibitors to block HDAC activity, we demonstrate that
- class I HDACs are key regulators of FoxO and the muscle-atrophy program
- during both nutrient deprivation and skeletal muscle disuse.
Furthermore, we demonstrate, through the use of wild-type and dominant-negative HDAC1 expression plasmids,
- that HDAC1 is sufficient to activate FoxO and induce muscle fiber atrophy in vivo and
- is necessary for the atrophy of muscle fibers that is associated with muscle disuse.
The ability of HDAC1 to cause muscle atrophy required its deacetylase activity and
- was linked to the induction of several atrophy genes by HDAC1,
- including atrogin-1, which required deacetylation of FoxO3a.
Moreover, pharmacological inhibition of class I HDACs during muscle disuse, using MS-275,
- significantly attenuated both disuse muscle fiber atrophy and contractile dysfunction.
Together, these data solidify the importance of class I HDACs in the muscle atrophy program and
- indicate that class I HDAC inhibitors are feasible countermeasures to impede muscle atrophy and weakness.
Autophagy and thyroid carcinogenesis: genetic and epigenetic links
F Morani, R Titone, L Pagano, et al. Endocr Relat Cancer Feb 1, 2014 21 R13-R29
http://dx.doi.org:/10.1530/ERC-13-0271
Autophagy is a vesicular process for the lysosomal degradation of protein aggregates and
- of damaged or redundant organelles.
Autophagy plays an important role in cell homeostasis, and there is evidence that
- this process is dysregulated in cancer cells.
Recent in vitro preclinical studies have indicated that autophagy is
- involved in the cytotoxic response to chemotherapeutics in thyroid cancer cells.
Indeed, several oncogenes and oncosuppressor genes implicated in thyroid carcinogenesis
- also play a role in the regulation of autophagy.
In addition, some epigenetic modulators involved in thyroid carcinogenesis also influence autophagy. In this review, we highlight the genetic and epigenetic factors that
- mechanistically link thyroid carcinogenesis and autophagy, thus substantiating the rationale for
- an autophagy-targeted therapy of aggressive and radio-chemo-resistant thyroid cancers.
Chapter 6: Proteomics
Introduction to Proteomics
Author and Curator: Larry H. Bernstein, MD, FCAP
We have had a considerable extended discussion of preoteins and peptides, protein sinthesis, amino acid incorporation into protein, and metabolism of carbohydrates and lipids. It is also clear that the historic practice of medicine, and the classification of biological systems has been highly dependent on the observations related to the observed phenotypical traits and disturbances of normal function that could be measured by traditional metabolic pathways for over a century.
What did we gain from the genomic revolution?
- Traceability of protein expression to a basic coded message
- The possibility of tracing disturbed cellular function to mutation related loss-of-function
- The ability to trace generational traits over long periods of time
- The promise of regenerating the enterprise of pharmacology and pharmaceutical intervention based on the silencing of or readjustment of regulated metabolic pathways to bring an adaptive rebalancing favoring extended life
What can we expect as we progress further as a result of the last two decades?
- There is a huge amount of information, as well as missing information that is necessary for adequately tackling the mastery of the life processes.
- There is a complex web of knowledge that goes beyond the genome and the one-gene one-enzyme, and the DNA-RNA-protein hypotheses that can only be realized by more full disclosure of the many metabolic control circuits involved in cellular homeostasis and adaptive control.
- The ability to come to disclosure and understanding of this cellular balancing will require the comprehensive exploration of the proteome and the active role of proteins and peptides in the functioning of all cells, and the organism.
- Proteomics will open up the discovery of new approaches to diagnostics and pharmaceutical discovery.
What about proteins? What can proteins do? What can’t they do!
- Enzymes are proteins that make sure that chemical reactions in your body take place up to a million times faster than they would without enzymes.
- Antibodies are proteins that help your immune system to fight disease.
- When you get an injury, the bleeding stops because of blood clots, thanks to the proteins fibrinogen and thrombin.
- Transport! Some proteins carry vitamins ot hormones from one place to another, or form tunnels (pores) in cell membranes that will let only specific molecules (or ions) through. Hemoglobin, a protein in your blood, carries oxygen from your lungs to your cells.
- Strength and support! Other proteins like collagen and keratin are strong and tough and make up your skin, hair, and fingernails. Collagen also supports your cells and organs so they don’t slosh around.
- Motion! The proteins myosin and actin make up much of your muscle tissue. They work together so your muscles can move you around. Some bacteria have cilia and flagella made out of proteins. The bacteria can whip these around to move from place to place.
http://www.pslc.ws/macrog/kidsmac/protein.htm
Proteins (/ˈproʊˌtiːnz/ or /ˈproʊti.ɨnz/) are large biological molecules, ormacromolecules,
- consisting of one or more long chains of amino acid residues.
Proteins perform a vast array of functions within living organisms, including
- catalyzing metabolic reactions,
- replicating DNA,
- responding to stimuli, and
- transporting molecules from one location to another.
Proteins differ from one another primarily in
- their sequence of amino acids,
- which is dictated by the nucleotide sequence of their genes, and
- which usually results in folding of the protein into
- a specific three-dimensional structure that determines its activity.
A linear chain of amino acid residues is called a polypeptide. A protein contains at least one long polypeptide. Short polypeptides, containing less than about 20-30 residues, are rarely considered to be proteins and are commonly called peptides, or sometimes oligopeptides. The individual amino acid residues are bonded together bypeptide bonds and adjacent amino acid residues. The sequence of amino acid residues in a protein is defined by
- the sequence of a gene, which is encoded in the genetic code.
In general, the genetic code specifies 20 standard amino acids; however, in certain organisms the genetic code can include selenocysteine and—in certainarchaea—pyrrolysine. Shortly after or even during synthesis,
- the residues in a protein are often chemically modified by posttranslational modification,
- which alters the physical and chemical properties, folding, stability, activity, and ultimately, the function of the proteins.
http://en.wikipedia.org/wiki/Protein
Posttranslational modification (PTM) is a step in protein biosynthesis. Proteins created by ribosomes translating mRNA into polypeptide chains may undergo PTM (such as folding, cutting and other processes) before becoming the mature protein product. After translation, the posttranslational modification of amino acids extends the range of functions of the protein by attaching it to other biochemical functional groups (such as acetate, phosphate, various lipids and carbohydrates), changing the chemical nature of an amino acid (e.g. citrullination), or making structural changes (e.g. formation of disulfide bridges).
Also, enzymes may remove amino acids from the amino end of the protein, or cut the peptide chain in the middle. For instance, the peptide hormone insulin is cut twice after disulfide bonds are formed, and a propeptide is removed from the middle of the chain; the resulting protein consists of two polypeptide chains connected by disulfide bonds. Also, most nascent polypeptides start with the amino acidmethionine because the “start” n mRNA also codes for this amino acid. This amino acid is usually taken off during post-translational modification. Other modifications, like phosphorylation, are part of common mechanisms for controlling the behavior of a protein, for instance activating or inactivating an enzyme.

Posttranslational modification of insulin. At the top, the ribosome translates a mRNA sequence into a protein, insulin, and passes the protein through the endoplasmic reticulum, where it is cut, folded and held in shape by disulfide (-S-S-) bonds. Then the protein passes through the golgi apparatus, where it is packaged into a vesicle. In the vesicle, more parts are cut off, and it turns into mature insulin.

The genetic code diagram showing the amino acid residues as target of modification.
PTMs involving addition of cofactors for enhanced enzymatic activity
- lipoylation, attachment of a lipoate (C8) functional group
- flavin moiety (FMN or FAD) may be covalently attached
- heme C attachment via thioether bonds with cysteins
- phosphopantetheinylation, the addition of a 4′-phosphopantetheinyl moiety from coenzyme A, as in fatty acid, polyketide, non-ribosomal peptide and leucine biosynthesis
- retinylidene Schiff base formation
http://en.wikipedia.org/wiki/Posttranslational_modification
Sometimes proteins have non-peptide groups attached, which can be calledprosthetic groups or cofactors. Examples of cofactors include metal ions like iron and zinc. Proteins can also work together to achieve a particular function, and they often associate to form stable protein complexes.

Coenzymes are molecules that work at the active site of an enzyme and aid in recognizing, attracting, or repulsing a substrate or product. Many are derived from vitamins. The substrate is the molecule upon which an enzyme catalyzes a reaction transforming A to B by removal or addition of a hydrogen, or a hydroxyl group, or a methyl group, and so forth. This is how an alcohol or an aldehyde is produced. Such a reaction is critical is carbohydrate metabolism for producing two 3-carbon sugars from a 6-carbon sugar. Coenzymes shuttle chemical groups from one enzyme to another enzyme. They may bind loosely to enzymes, while another group of cofactors do not.
Prosthetic groups are cofactors that bind tightly to proteins or enzymes. As if holding on for dear life, they are not easily removed. They can be organic or metal ions and are often attached to proteins by a covalent bond. The same cofactors can bind multiple different types of enzymes and may bind some enzymes loosely, as a coenzyme, and others tightly, as a prosthetic group. Some cofactors may always tightly bind their enzymes. It’s important to note, though, that these prosthetic groups can also bind to proteins other than enzymes. A holoenzyme is an enzyme with any metal ions or coenzymes attached to it that is now ready to catalyze a reaction.

Around the world, millions of people don’t get enough protein. Protein malnutrition leads to the condition known as kwashiorkor. Lack of protein can cause growth failure, loss of muscle mass, decreased immunity, weakening of the heart and respiratory system, and death.
All Protein Isn’t Alike
Protein is built from building blocks called amino acids. Our bodies make amino acids in two different ways: Either from scratch, or by modifying others. A few amino acids (known as the essential amino acids) must come from food.
- Animal sources of protein tend to deliver all the amino acids we need.
- Other protein sources, such as fruits, vegetables, grains, nuts and seeds, lack one or more essential amino acids.
Vegetarians need to be aware of this. People who don’t eat meat, fish, poultry, eggs, or dairy products need to eat a variety of protein-containing foods each day in order to get all the amino acids needed to make new protein.
http://www.hsph.harvard.edu/nutritionsource/what-should-you-eat/protein/
Molecular Biologists Guide to Proteomics
PR. Graves and TA.J. Haystead*
Microbiol Mol Biol Rev. Mar 2002; 66(1): 39–63 PMC120780
http://dx.doi.org:/10.1128/MMBR.66.1.39-63.2002
The emergence of proteomics, the large-scale analysis of proteins, has been inspired by the realization that
- the final product of a gene is inherently more complex and
- closer to function than the gene itself.
Shortfalls in the ability of bioinformatics to predict
- both the existence and function of genes have also illustrated
- the need for protein analysis.
Moreover, only through the study of proteins can posttranslational modifications be determined,
- which can profoundly affect protein function.
Proteomics has been enabled by
- the accumulation of both DNA and protein sequence databases,
- improvements in mass spectrometry, and
- the development of computer algorithms for database searching.
In this review, we describe why proteomics is important,
- how it is conducted, and
- how it can be applied to complement other existing technologies.
We conclude that currently, the most practical application of proteomics is
- the analysis of target proteins as opposed to entire proteomes.
This type of proteomics, referred to as functional proteomics, is always
- driven by a specific biological question.
In this way, protein identification and characterization has a meaningful outcome. We discuss some of the advantages
- of a functional proteomics approach and
provide examples of how different methodologies can be utilized to address a wide variety of biological problems.
Entry of our laboratory into proteomics 5 years ago was driven by a need to define a complex mixture of proteins (∼36 proteins) we had affinity isolated that bound specifically to the catalytic subunit of protein phosphatase 1 (PP-1, a serine/threonine protein phosphatase that regulates multiple dephosphorylation events in cells). We were faced with the task of trying to understand the significance of these proteins, and the only obvious way to begin to do this was to identify them by sequencing. Since the majority of intact eukaryotic proteins are not immediately accessible to Edman sequencing
- due to posttranslational N-terminal modifications,
- we invented mixed-peptide sequencing.
This method enables internal peptide sequence information to be derived from proteins
- electroblotted onto hydrophobic membranes.
Using the mixed-peptide sequencing strategy, we identified all 36 proteins in about a week. The mixture contained at least two known PP-1 regulatory subunits, but most were novel proteins of unknown function. Herein lies the lesson of proteomics. Identifying long lists of potentially interesting proteins often generates more questions than it seeks to answer.
Despite learning this obvious lesson, our early sequencing experiences were an epiphany that has subsequently altered our whole scientific strategy for probing protein function in cells. The sequencing of the 36 proteins has opened new avenues to further explore the functions of PP-1 in intact cells. Because of increased sensitivity, our approaches now routinely use state-of-the-art mass spectrometry (MS) techniques. However, rather than using proteomics to simply characterize large numbers of proteins in complex mixtures, we see the real application of this technology as a tool to enhance the power of existing approaches currently used by the modern molecular biologist such as classical yeast and mouse genetics, tissue culture, protein expression systems, and site-directed mutagenesis.
Importantly, the one message we would want the reader to take away from reading this review is that one should always let the biological question in mind drive the application of proteomics rather than simply engaging in an orgy of protein sequencing. From our experiences, we believe that if the appropriate controls are performed, proteomics is an extremely powerful approach for addressing important physiological questions. One should always design experiments to define a selected number of relevant proteins in the mixture of interest. Examples of such experiments that we routinely perform include defining early phosphorylation events in complex protein mixtures after hormone treatment of intact cells or comparing patterns of protein derived from a stimulated versus nonstimulated cell in an affinity pull-down experiment. Only the proteins that were specifically phosphorylated or bound in response to the stimulus are sequenced in the complex mixtures. Sequencing proteins that are regulated then has a meaningful outcome and directs all subsequent biological investigation.
The term “proteomics” was first coined in 1995 and was defined as the large-scale characterization of the entire protein complement of a cell line, tissue, or organism. Today, two definitions of proteomics are encountered. The first is the more classical definition, restricting the large-scale analysis of gene products to studies involving only proteins. The second and more inclusive definition combines protein studies with analyses that have a genetic readout such as mRNA analysis, genomics, and the yeast two-hybrid analysis. However, the goal of proteomics remains the same, i.e., to obtain a more global and integrated view of biology by studying all the proteins of a cell rather than each one individually.
Using the more inclusive definition of proteomics, many different areas of study are now grouped under the rubric of proteomics (Fig. (Fig.1).1). These include protein-protein interaction studies, protein modifications, protein function, and protein localization studies to name a few. The aim of proteomics is not only to identify all the proteins in a cell but also to create a complete three-dimensional (3-D) map of the cell indicating where proteins are located. These ambitious goals will certainly require the involvement of a large number of different disciplines such as molecular biology, biochemistry, and bioinformatics. It is likely that in bioinformatics alone, more powerful computers will have to be devised to organize the immense amount of information generated from these endeavors.

In the quest to characterize the proteome of a given cell or organism, it should be remembered that the proteome is dynamic. The proteome of a cell will reflect the immediate environment in which it is studied. In response to internal or external cues, proteins can be modified by posttranslational modifications, undergo translocations within the cell, or be synthesized or degraded. Thus, examination of the proteome of a cell is like taking a “snapshot” of the protein environment at any given time. Considering all the possibilities, it is likely that any given genome can potentially give rise to an infinite number of proteomes.
The first major technology to emerge for the identification of proteins was the sequencing of proteins by Edman degradation. A major breakthrough was the development of microsequencing techniques for electroblotted proteins. This technique was used for the identification of proteins from 2-D gels to create the first 2-D databases. One of the most important developments in protein identification has been the development of MS technology. In the last decade, the sensitivity of analysis and accuracy of results for protein identification by MS have increased by several orders of magnitude. It is now estimated that proteins in the femtomolar range can be identified in gels. Because MS is more sensitive, can tolerate protein mixtures, and is amenable to high-throughput operations, it has essentially replaced Edman sequencing as the protein identification tool of choice.
The growth of proteomics is a direct result of advances made in large-scale nucleotide sequencing of expressed sequence tags and genomic DNA. Without this information, proteins could not be identified even with the improvements made in MS. Protein identification (by MS or Edman sequencing) relies on the presence of some form of database for the given organism. The majority of DNA and protein sequence information has accumulated within the last 5 to 10 years. In 1995, the first complete genome of an organism was sequenced, that of Haemophilus influenzae. At the time of this writing, the sequencing of the genomes of 45 microorganisms has been completed and that of 170 more is under way (http://www.tiger.org/tdb/mdb/mdbcomplete.html). To date, five eukaryotic genomes have been completed: Arabidopsis thaliana, Saccharomyces cerevisiae,Schizosaccharomyces pombe, Caenorhabditis elegans, and Drosophila melanogaster. In addition, the rice, mouse, and human genomes are near completion.
One of the first applications of proteomics will be to identify the total number of genes in a given genome. This “functional annotation” of a genome is necessary because
- it is still difficult to predict genes accurately from genomic data. One problem is that
- the exon-intron structure of most genes cannot be accurately predicted by bioinformatics.
To achieve this goal, genomic information will have to be integrated with
- data obtained from protein studies to confirm the existence of a particular gene.
The analysis of mRNA is
- not a direct reflection of the protein content in the cell.
Many studies have shown a poor correlation
- between mRNA and protein expression levels.
The formation of mRNA is only the first step in a long sequence of events resulting in the synthesis of a protein (Fig. (Fig.2).2).
- mRNA is subject to posttranscriptional control in the form of alternative splicing, polyadenylation, and mRNA editing. Many different protein isoforms can be generated from a single gene at this step.
- mRNA then can be subject to regulation at the level of protein translation. Proteins, having been formed, are subject toposttranslational modification. It is estimated that up to 200 different types of posttranslational protein modification exist. Proteins can also be regulated by proteolysis and compartmentalization. It is clear that the tenet of “one gene, one protein” is an oversimplification.




Triple-quadrupole mass spectrometers are most commonly used to obtain amino acid sequences. In the first stage of analysis, the machine is operated in MS scan mode and all ions above a certain m/z ratio are transmitted to the third quadrupole for mass analysis (Fig. (Fig.6)6) (82, 173). In the second stage, the mass spectrometer is operated in MS/MS mode and a particular peptide ion is selectively passed into the collision chamber. Inside the collision chamber, peptide ions are fragmented by interactions with an inert gas by a process known as collision-induced dissociation or collisionally activated dissociation. The peptide ion fragments are then resolved on the basis of their m/z ratio by the third quadrupole (Fig. (Fig.6).6). Since two different mass spectra are obtained in this analysis, it is referred to as tandem mass spectrometry (MS/MS). MS/MS is used to obtain the amino acid sequence of peptides by generating a series of peptides that differ in mass by a single amino acid.
The largest application of proteomics continues to be protein expression profiling. Through the use of two-dimensional gels or novel techniques such as ICAT, the expression levels of proteins or changes in their level of modification between two different samples can be compared and the proteins can be identified. This approach can facilitate the dissection of signaling mechanisms or identify disease-specific proteins.
Cancer cells are good candidates for proteomics studies because they can be compared to their non-transformed counterparts. Analysis of differentially expressed proteins in normal versus cancer cells can
(i) identify novel tumor cell biomarkers that can be used for diagnosis,
(ii) provide clues to mechanisms of cancer development, and
(iii) identify novel targets for therapeutic intervention. Protein expression profiling has been used in the study of breast, esophageal, bladder and prostate cancer. From these studies, tumor-specific proteins were identified and 2-D protein expression databases were generated. Many of these 2-D protein databases are now available on the World Wide Web.
Summary of Proteomics
Author and Curator: Larry H. Bernstein, MD, FCAP
We have completed a series of discussions on proteomics, a scientific endeavor that is essentially 15 years old. It is quite remarkable what has been accomplished in that time. The interest is abetted by the understanding of the limitations of the genomic venture that has preceded it. The thorough, yet incomplete knowledge of the genome, has led to the clarification of its limits. It is the coding for all that lives, but all that lives has evolved to meet a demanding and changing environment with respect to
- availability of nutrients
- salinity
- temperature
- radiation exposure
- toxicities in the air, water, and food
- stresses – both internal and external
We have seen how both transcription and translation of the code results in a protein, lipoprotein, or other complex than the initial transcript that was modeled from tRNA. What you see in the DNA is not what you get in the functioning cell, organ, or organism. There are comparabilities as well as significant differences between plants, prokaryotes, and eukaryotes. There is extensive variation. The variation goes beyond genomic expression, and includes the functioning cell, organ type, and species.
Here, I return to the introductory discussion. Proteomics is a goal directed, sophisticated science that uses a combination of methods to find the answers to biological questions. Graves PR and Haystead TAJ. Molecular Biologist’s Guide to Proteomics.
Microbiol Mol Biol Rev. Mar 2002; 66(1): 39–63. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC120780/

Peptide mass tag searching. Shown is a schematic of how information from an unknown peptide (top) is matched to a peptide sequence in a database (bottom) for protein identification. The partial amino acid sequence or “tag” obtained by MS/MS is combined with the peptide mass (parent mass), the mass of the peptide at the start of the sequence (mass tag 1), and the mass of the peptide at the end of the sequence (mass tag 2). The specificity of the protease used (trypsin is shown) can also be included in the search.

The ICAT method for measuring differential protein expression. (A) Structure of the ICAT reagent. ICAT consists of a biotin affinity group, a linker region that can incorporate heavy (deuterium) or light (hydrogen) atoms, and a thiol-reactive end group for linkage to cysteines. (B) ICAT strategy. Proteins are harvested from two different cell states and labeled on cysteine residues with either the light or heavy form of the ICAT reagent. Following labeling, the two protein samples are mixed and digested with a protease such as trypsin. Peptides labeled with the ICAT reagent can be purified by virtue of the biotin tag by using avidin chromatography. Following purification, ICAT-labeled peptides can be analyzed by MS to quantitate the peak ratios and proteins can be identified by sequencing the peptides with MS/MS.

Strategies for determination of phosphorylation sites in proteins. Proteins phosphorylated in vitro or in vivo can be isolated by protein electrophoresis and analyzed by MS. (A) Identification of phosphopeptides by peptide mass fingerprinting. In this method, phosphopeptides are identified by comparing the mass spectrum of an untreated sample to that of a sample treated with phosphatase. In the phosphatase-treated sample, potential phosphopeptides are identified by a decrease in mass due to loss of a phosphate group (80 Da). (B) Phosphorylation sites can be identified by peptide sequencing using MS/MS. (C) Edman degradation can be used to monitor the release of inorganic 32P to provide information about phosphorylation sites in peptides.

Proteome-mining strategy. Proteins are isolated on affinity column arrays from a cell line, organ, or animal source and purified to remove nonspecific adherents. Then, compound libraries are passed over the array and the proteins eluted are analyzed by protein electrophoresis. Protein information obtained by MS or Edman degradation is then used to search DNA and protein databases. If a relevant target is identified, a sublibrary of compounds can be evaluated to refine the lead. From this method a protein target and a drug lead can be simultaneously identified.
Although the technology for the analysis of proteins is rapidly progressing, it is still not feasible to study proteins on a scale equivalent to that of the nucleic acids. Most of proteomics relies on methods, such as protein purification or PAGE, that are not high-throughput methods. Even performing MS can require considerable time in either data acquisition or analysis. Although hundreds of proteins can be analyzed quickly and in an automated fashion by a MALDI-TOF mass spectrometer, the quality of data is sacrificed and many proteins cannot be identified. Much higher quality data can be obtained for protein identification by MS/MS, but this method requires considerable time in data interpretation. In our opinion, new computer algorithms are needed to allow more accurate interpretation of mass spectra without operator intervention. In addition, to access unannotated DNA databases across species, these algorithms should be error tolerant to allow for sequencing errors, polymorphisms, and conservative substitutions. New technologies will have to emerge before protein analysis on a large-scale (such as mapping the human proteome) becomes a reality.
Another major challenge for proteomics is the study of low-abundance proteins. In some eukaryotic cells, the amounts of the most abundant proteins can be 106-fold greater than those of the low-abundance proteins. Many important classes of proteins (that may be important drug targets) such as transcription factors, protein kinases, and regulatory proteins are low-copy proteins. These low-copy proteins will not be observed in the analysis of crude cell lysates without some purification. Therefore, new methods must be devised for subproteome isolation.
Tissue Proteomics for the Next Decade? Towards a Molecular Dimension in Histology
R Longuespe´e, M Fle´ron, C Pottier, F Quesada-Calvo, Marie-Alice Meuwis, et al.
OMICS A Journal of Integrative Biology 2014; 18: 9. http://dx.doi.org:/10.1089/omi.2014.0033
The concept of tissues appeared more than 200 years ago, since textures and attendant differences were described within the whole organism components. Instrumental developments in optics and biochemistry subsequently paved the way to transition from classical to molecular histology in order to decipher the molecular contexts associated with physiological or pathological development or function of a tissue. In 1941, Coons and colleagues performed the first systematic integrated examination of classical histology and biochemistry when his team localized pneumonia antigens in infected tissue sections. Most recently, in the early 21st century, mass spectrometry (MS) has progressively become one of the most valuable tools to analyze biomolecular compounds. Currently, sampling methods, biochemical procedures, and MS instrumentations
allow scientists to perform ‘‘in depth’’ analysis of the protein content of any type of tissue of interest. This article reviews the salient issues in proteomics analysis of tissues. We first outline technical and analytical considerations for sampling and biochemical processing of tissues and subsequently the instrumental possibilities for proteomics analysis such as shotgun proteomics in an anatomical context. Specific attention concerns formalin fixed and paraffin embedded (FFPE) tissues that are potential ‘‘gold mines’’ for histopathological investigations. In all, the matrix assisted laser desorption/ionization (MALDI) MS imaging, which allows for differential mapping of hundreds of compounds on a tissue section, is currently the most striking evidence of linkage and transition between ‘‘classical’’ and ‘‘molecular’’ histology. Tissue proteomics represents a veritable field of research and investment activity for modern biomarker discovery and development for the next decade.
Progressively, tissue analyses evolved towards the description of the whole molecular content of a given sample. Currently, mass spectrometry (MS) is the most versatile
analytical tool for protein identification and has proven its great potential for biological and clinical applications. ‘‘Omics’’ fields, and especially proteomics, are of particular
interest since they allow the analysis of a biomolecular picture associated with a given physiological or pathological state. Biochemical techniques were then adapted for an optimal extraction of several biocompounds classes from tissues of different natures.
Laser capture microdissection (LCM) is used to select and isolate tissue areas of interest for further analysis. The developments of MS instrumentations have then definitively transformed the scientific scene, pushing back more and more detection and identification limits. Since a few decades, new approaches of analyses appeared, involving the use of tissue sections dropped on glass slides as starting material. Two types of analyses can then be applied on tissue sections: shotgun proteomics and the very promising MS imaging (MSI) using Matrix Assisted Laser Desorption/Ionization (MALDI) sources. Also known as ‘‘molecular histology,’’ MSI is the most striking hyphen between histology and molecular analysis. In practice, this method allows visualization of the spatial distribution of proteins, peptides, drugs, or others analytes directly on tissue sections. This technique paved new ways of research, especially in the field of histopathology, since this approach appeared to be complementary to conventional histology.

Tissue processing workflows for molecular analyses. Tissues can either be processed in solution or directly on tissue sections. In solution, processing involves protein extraction from tissue pieces in order to perform 2D gel separation and identification of proteins, shotgun proteomics, or MALDI analyses. Extracts can also be obtained from tissues area selection and protein extraction after laser micro dissection or on-tissue processing. Imaging techniques are dedicated to the morphological characterization or molecular mapping of tissue sections. Histology can either be conducted by hematoxylin/eosin staining or by molecular mapping using antibodies with IHC. Finally, mass spectrometry imaging allows the cartography of numerous compounds in a single analysis. This approach is a modern form of ‘‘molecular histology’’ as it grafts, with the use of mathematical calculations, a molecular dimension to classical histology. (AR, antigen retrieval; FFPE, formalin fixed and paraffin embedded; fr/fr, fresh frozen; IHC, immunohistochemistry; LCM, laser capture microdissection; MALDI, matrix assisted laser desorption/ionization; MSI, mass spectrometry imaging; PTM, post translational modification.)
Analysis of tissue proteomes has greatly evolved with separation methods and mass spectrometry instrumentation. The choice of the workflow strongly depends on whether a bottom-up or a top-down analysis has to be performed downstream. In-gel or off-gel proteomics principally differentiates proteomic workflows. The almost simultaneous discoveries of the MS ionization sources (Nobel Prize awarded) MALDI (Hillenkamp and Karas, 1990; Tanaka et al., 1988) and electrospray ionization (ESI) (Fenn et al., 1989) have paved the way for analysis of intact proteins and peptides. Separation methods such as two-dimension electrophoresis (2DE) (Fey and Larsen, 2001) and nanoscale reverse phase liquid chromatography (nanoRP-LC) (Deterding et al., 1991) lead to efficient preparation of proteins for respectively topdown and bottom-up strategies. A huge panel of developments was then achieved mostly for LC-MS based proteomics in order to improve ion fragmentation approaches and peptide identification throughput relying on database interrogation. Moreover, approaches were developed to analyze post translational modifications (PTM) such as phosphorylations (Ficarro et al., 2002; Oda et al., 2001; Zhou et al., 2001) or glycosylations (Zhang et al., 2003), proposing as well different quantification procedures. Regarding instrumentation, the most cutting edge improvements are the gain of mass accuracy for an optimal detection of the eluted peptides during LC-MS runs (Mann and Kelleher, 2008; Michalski et al., 2011) and the increase in scanning speed, for example with the use of Orbitrap analyzers (Hardman and Makarov, 2003; Makarov et al., 2006; Makarov et al., 2009; Olsen et al., 2009). Ion transfer efficiency was also drastically improved with the conception of ion funnels that homogenize the ion transmission capacities through m/z ranges (Kelly et al., 2010; Kim et al., 2000; Page et al., 2006; Shaffer et al., 1998) or by performing electrospray ionization within low vacuum (Marginean et al., 2010; Page et al., 2008; Tang et al., 2011). Beside collision induced dissociation (CID) that is proposed for many applications (Li et al., 2009; Wells and McLuckey, 2005), new fragmentation methods were investigated, such as higher-energy collisional dissociation (HCD) especially for phosphoproteomic applications (Nagaraj et al., 2010), and electron transfer dissociation (ETD) and electron capture dissociation (ECD) that are suited for phospho- and glycoproteomics (An et al., 2009; Boersema et al., 2009; Wiesner et al., 2008). Methods for data-independent MS2 analysis based on peptide fragmentation in given m/z windows without precursor selection neither information knowledge, also improves identification throughput (Panchaud et al., 2009; Venable et al., 2004), especially with the use of MS instruments with high resolution and high mass accuracy specifications (Panchaud et al., 2011). Gas fractionation methods such as ion mobility (IM) can also be used as a supplementary separation dimension which enable more efficient peptide identifications (Masselon et al., 2000; Shvartsburg et al., 2013; Shvartsburg et al., 2011).
Microdissection relies on a laser ablation principle. The tissue section is dropped on a plastic membrane covering a glass slide. The preparation is then placed into a microscope equipped with a laser. A highly focused beam will then be guided by the user at the external limit of the area of interest. This area composed by the plastic membrane, and the tissue section will then be ejected from the glass slide and collected into a tube cap for further processing. This mode of microdissection is the most widely used due to its ease of handling and the large panels of devices proposed by constructors. Indeed, Leica microsystem proposed the Leica LMD system (Kolble, 2000), Molecular Machine and Industries, the MMI laser microdissection system Microcut, which was used in combination with IHC (Buckanovich et al., 2006), Applied Biosystems developed the Arcturus microdissection System, and Carl Zeiss patented P.A.L.M. MicroBeam technology (Braakman et al., 2011; Espina et al., 2006a; Espina et al., 2006b; Liu et al., 2012; Micke et al., 2005). LCM represents a very adequate link between classical histology and sampling methods for molecular analyses as it is a simple customized microscope. Indeed,
optical lenses of different magnification can be used and the method is compatible with classical IHC (Buckanovich et al., 2006). Only the laser and the tube holder need to be added to the instrumentation.
After microdissection, the tissue pieces can be processed for analyses using different available MS devices and strategies. The simplest one consists in the direct analysis of the protein profiles by MALDI-TOF-MS (MALDI-time of flight-MS). The microdissected tissues are dropped on a MALDI target and directly covered by the MALDI matrix (Palmer-Toy et al., 2000; Xu et al., 2002). This approach was already used in order to classify breast cancer tumor types (Sanders et al., 2008), identify intestinal neoplasia protein biomarkers (Xu et al., 2009), and to determine differential profiles in glomerulosclerosis (Xu et al., 2005).
Currently the most common proteomic approach for LCM tissue analysis is LC-MS/MS. Label free LC-MS approaches have been used to study several cancers like head and neck squamous cell carcinomas (Baker et al., 2005), esophageal cancer (Hatakeyama et al., 2006), dysplasic cervical cells (Gu et al., 2007), breast carcinoma tumors (Hill et al., 2011; Johann et al., 2009), tamoxifen-resistant breast cancer cells (Umar et al., 2009), ER + / – breast cancer cells (Rezaul et al., 2010), Barretts esophagus (Stingl et al., 2011), and ovarian endometrioid cancer (Alkhas et al., 2011). Different isotope labeling methods have been used in order to compare proteins expression. ICAT was first used to investigate proteomes of hepatocellular carcinoma (Li et al., 2004; 2008). The O16/O18 isotopic labeling was then used for proteomic analysis of ductal carcinoma of the breast (Zang et al., 2004).
Currently, the lowest amount of collected cells for a relevant single analysis using fr/fr breast cancer tissues was 3000–4000 (Braakman et al., 2012; Liu et al., 2012; Umar et al., 2007). With a Q-Exactive (Thermo, Waltham) mass spectrometer coupled to LC, Braakman was able to identify up to 1800 proteins from 4000 cells. Processing of FFPE microdissected tissues of limited sizes still remains an issue which is being addressed by our team.
Among direct tissue analyses modes, two categories of investigations can be done. MALDI profiling consists in the study of molecular localization of compounds and can be combined with parallel shotgun proteomic methods. Imaging methods give less detailed molecular information, but is more focused on the accurate mapping of the detected compounds through tissue area. In 2007, a concept of direct tissue proteomics (DTP) was proposed for high-throughput examination of tissue microarray samples. However, contrary to the classical workflow, tissue section chemical treatment involved a first step of scrapping each FFPE tissue spot with a razor blade from the glass slide. The tissues were then transferred into a tube and processed with RIPA buffer and finally submitted to boiling as an AR step (Hwang et al., 2007). Afterward, several teams proved that it was possible to perform the AR directly on tissue sections. These applications were mainly dedicated to MALDI imaging analyses (Bonnel et al., 2011; Casadonte and Caprioli, 2011; Gustafsson et al., 2010). However, more recently, Longuespe´e used citric acid antigen retrieval (CAAR) before shotgun proteomics associated to global profiling proteomics (Longuespee et al., 2013).

MALDI imaging workflow. For MALDI imaging experiments, tissue sections are dropped on conductive glass slides. Sample preparations are then adapted depending on the nature of the tissue sample (FFPE or fr/fr). Then, matrix is uniformly deposited on the tissue section using dedicated devices. A laser beam subsequently irradiates the preparation following a given step length and a MALDI spectrum is acquired for each position. Using adapted software, the different detected ions are then mapped through the tissue section, in function of their differential intensities. The ‘‘molecular maps’’ are called images. (FFPE, formalin fixed and paraffin embedded; fr/fr, fresh frozen; MALDI, matrix assisted laser desorption ionization.)
Proteomics instrumentations, specific biochemical preparations, and sampling methods such as LCM altogether allow for the deep exploration and comparison of different proteomes between regions of interest in tissues with up to 104 detected proteins. MALDI MS imaging that allows for differential mapping of hundreds of compounds on a tissue section is currently the most striking illustration of association between ‘‘classical’’ and ‘‘molecular’’ histology.
Novel serum protein biomarker panel revealed by mass spectrometry and its prognostic value in breast cancer
L Chung, K Moore, L Phillips, FM Boyle, DJ Marsh and RC Baxter* Breast Cancer Research 2014, 16:R63
http://breast-cancer-research.com/content/16/3/R63
Introduction: Serum profiling using proteomic techniques has great potential to detect biomarkers that might improve diagnosis and predict outcome for breast cancer patients (BC). This study used surface-enhanced laser desorption/ionization time-of-flight (SELDI-TOF) mass spectrometry (MS) to identify differentially expressed proteins in sera from BC and healthy volunteers (HV), with the goal of developing a new prognostic biomarker panel.
Methods: Training set serum samples from 99 BC and 51 HV subjects were applied to four adsorptive chip surfaces (anion-exchange, cation-exchange, hydrophobic, and metal affinity) and analyzed by time-of-flight MS. For validation, 100 independent BC serum samples and 70 HV samples were analyzed similarly. Cluster analysis of protein spectra was performed to identify protein patterns related to BC and HV groups. Univariate and multivariate statistical analyses were used to develop a protein panel to distinguish breast cancer sera from healthy sera, and its prognostic potential was evaluated.
Results: From 51 protein peaks that were significantly up- or downregulated in BC patients by univariate analysis, binary logistic regression yielded five protein peaks that together classified BC and HV with a receiver operating characteristic (ROC) area-under-the-curve value of 0.961. Validation on an independent patient cohort confirmed the five-protein parameter (ROC value 0.939). The five-protein parameter showed positive association with large tumor size (P = 0.018) and lymph node involvement (P = 0.016). By matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS, immunoprecipitation and western blotting the proteins were identified as a fragment of apolipoprotein H (ApoH), ApoCI, complement C3a, transthyretin, and ApoAI. Kaplan-Meier analysis on 181 subjects after median follow-up of >5 years demonstrated that the panel significantly predicted disease-free survival (P = 0.005), its efficacy apparently greater in women with estrogen receptor (ER)-negative tumors (n = 50, P = 0.003) compared to ER-positive (n = 131, P = 0.161), although the influence of ER status needs to be confirmed after longer follow-up.
Conclusions: Protein mass profiling by MS has revealed five serum proteins which, in combination, can distinguish between serum from women with breast cancer and healthy control subjects with high sensitivity and specificity. The five-protein panel significantly predicts recurrence-free survival in women with ER-negative tumors and may have value in the management of these patients.
Cellular prion protein is required for neuritogenesis: fine-tuning of multiple signaling pathways involved in focal adhesions and actin cytoskeleton dynamics
Aurélie Alleaume-Butaux, et al. Cell Health and Cytoskeleton 2013:5 1–12
Neuritogenesis is a dynamic phenomenon associated with neuronal differentiation that allows a rather spherical neuronal stem cell to develop dendrites and axon, a prerequisite for the integration and transmission of signals. The acquisition of neuronal polarity occurs in three steps:
(1) neurite sprouting, which consists of the formation of buds emerging from the postmitotic neuronal soma;
(2) neurite outgrowth, which represents the conversion of buds into neurites, their elongation and evolution into axon or dendrites; and
(3) the stability and plasticity of neuronal polarity.
In neuronal stem cells, remodeling and activation of focal adhesions (FAs)
- associated with deep modifications of the actin cytoskeleton is
- a prerequisite for neurite sprouting and subsequent neurite outgrowth.
A multiple set of growth factors and interactors located in
- the extracellular matrix and the plasma membrane orchestrate neuritogenesis
- by acting on intracellular signaling effectors, notably small G proteins such as RhoA, Rac, and Cdc42,
- which are involved in actin turnover and the dynamics of FAs.
The cellular prion protein (PrPC), a glycosylphosphatidylinositol (GPI)-anchored membrane protein
- mainly known for its role in a group of fatal neurodegenerative diseases,
- has emerged as a central player in neuritogenesis.
Here, we review the contribution of PrPC to neuronal polarization and
- detail the current knowledge on the signaling pathways fine-tuned
- by PrPC to promote neurite sprouting, outgrowth, and maintenance.
We emphasize that PrPC-dependent neurite sprouting is a process in which
- PrPC governs the dynamics of FAs and the actin cytoskeleton via β1 integrin signaling.
The presence of PrPC is necessary to render neuronal stem cells
- competent to respond to neuronal inducers and to develop neurites.
In differentiating neurons, PrPC exerts a facilitator role towards neurite elongation.
This function relies on the interaction of PrPC with a set of diverse partners such as
- elements of the extracellular matrix,
- plasma membrane receptors,
- adhesion molecules, and
- soluble factors that control actin cytoskeleton turnover
- through Rho-GTPase signaling.
Once neurons have reached their terminal stage of differentiation and
- acquired their polarized morphology,
- PrPC also takes part in the maintenance of neurites.
By acting on tissue nonspecific alkaline phosphatase, or matrix metalloproteinase type 9,
- PrPC stabilizes interactions between neurites and the extracellular matrix.
Fusion-pore expansion during syncytium formation is restricted by an actin network
Andrew Chen et al., Journal of Cell Science 121, 3619-3628.http://dx.doi.org:/10.1242/jcs.032169
Cell-cell fusion in animal development and in pathophysiology
- involves expansion of nascent fusion pores formed by protein fusogens
- to yield an open lumen of cell-size diameter.
Here we explored the enlargement of micron-scale pores in syncytium formation,
- which was initiated by a well-characterized fusogen baculovirus gp64.
Radial expansion of a single or, more often, of multiple fusion pores
- proceeds without loss of membrane material in the tight contact zone.
Pore growth requires cell metabolism and is
- accompanied by a local disassembly of the actin cortex under the pores.
Effects of actin-modifying agents indicate that
- the actin cortex slows down pore expansion.
We propose that the growth of the strongly bent fusion-pore rim
- is restricted by a dynamic resistance of the actin network and
- driven by membrane-bending proteins that are involved in
- the generation of highly curved intracellular membrane compartments.
Pak1 Is Required to Maintain Ventricular Ca2+ Homeostasis and Electrophysiological Stability Through SERCA2a Regulation in Mice
Yanwen Wang, et al. Circ Arrhythm Electrophysiol. 2014;7:00-00.
Impaired sarcoplasmic reticular Ca2+ uptake resulting from
- decreased sarcoplasmic reticulum Ca2+-ATPase type 2a (SERCA2a) expression or activity
- is a characteristic of heart failure with its associated ventricular arrhythmias.
Recent attempts at gene therapy of these conditions explored strategies
- enhancing SERCA2a expression and the activity as novel approaches to heart failure management.
We here explore the role of Pak1 in maintaining ventricular Ca2+ homeostasis and electrophysiological stability
- under both normal physiological and acute and chronic β-adrenergic stress conditions.
Methods and Results—Mice with a cardiomyocyte-specific Pak1 deletion (Pak1cko), but not controls (Pak1f/f), showed
- high incidences of ventricular arrhythmias and electrophysiological instability
- during either acute β-adrenergic or chronic β-adrenergic stress leading to hypertrophy,
- induced by isoproterenol.
Isolated Pak1cko ventricular myocytes correspondingly showed
- aberrant cellular Ca2+ homeostasis.
Pak1cko hearts showed an associated impairment of SERCA2a function and
- downregulation of SERCA2a mRNA and protein expression.
Further explorations of the mechanisms underlying the altered transcriptional regulation
- demonstrated that exposure to control Ad-shC2 virus infection
- increased SERCA2a protein and mRNA levels after
- phenylephrine stress in cultured neonatal rat cardiomyocytes.
This was abolished by the
- Pak1-knockdown in Ad-shPak1–infected neonatal rat cardiomyocytes and
- increased by constitutive overexpression of active Pak1 (Ad-CAPak1).
We then implicated activation of serum response factor, a transcriptional factor well known for
- its vital role in the regulation of cardiogenesis genes in the Pak1-dependent regulation of SERCA2a.
Conclusions—These findings indicate that
Pak1 is required to maintain ventricular Ca2+ homeostasis and electrophysiological stability
- and implicate Pak1 as a novel regulator of cardiac SERCA2a through
- a transcriptional mechanism
fusion in animal development and in pathophysiology involves expansion of nascent fusion pores
- formed by protein fusogens to yield an open lumen of cell-size diameter.
Here we explored the enlargement of micron-scale pores in syncytium formation,
- which was initiated by a well-characterized fusogen baculovirus gp64.
Radial expansion of a single or, more often, of multiple fusion pores proceeds
- without loss of membrane material in the tight contact zone.
Pore growth requires cell metabolism and is accompanied by
- a local disassembly of the actin cortex under the pores.
Effects of actin-modifying agents indicate that the actin cortex slows down pore expansion.
We propose that the growth of the strongly bent fusion-pore rim is restricted
- by a dynamic resistance of the actin network and driven by
- membrane-bending proteins that are involved in the generation of
- highly curved intracellular membrane compartments.
Role of forkhead box protein A3 in age-associated metabolic decline
Xinran Maa,1, Lingyan Xua,1, Oksana Gavrilovab, and Elisabetta Muellera,2
PNAS Sep 30, 2014 | 111 | 39 | 14289–14294 http://pnas.org/cgi/doi/10.1073/pnas.1407640111
Significance
This paper reports that the transcription factor forkhead box protein A3 (Foxa3) is
- directly involved in the development of age-associated obesity and insulin resistance.
Mice that lack the Foxa3 gene
- remodel their fat tissues,
- store less fat, and
- burn more energy as they age.
These mice also live significantly longer.
We show that Foxa3 suppresses a key metabolic cofactor, PGC1α,
- which is involved in the gene programs that turn on energy expenditure in adipose tissues.
Overall, these findings suggest that Foxa3 contributes to the increased adiposity observed during aging,
- and that it can be a possible target for the treatment of metabolic disorders.
Aging is associated with increased adiposity and diminished thermogenesis, but
- the critical transcription factors influencing these metabolic changes late in life are poorly understood.
We recently demonstrated that the winged helix factor forkhead box protein A3 (Foxa3)
- regulates the expansion of visceral adipose tissue in high-fat diet regimens; however,
- whether Foxa3 also contributes to the increase in adiposity and the decrease in brown fat activity
- observed during the normal aging process is currently unknown.
Here we report that during aging, levels of Foxa3 are significantly and selectively
- up-regulated in brown and inguinal white fat depots, and that
- midage Foxa3-null mice have increased white fat browning and thermogenic capacity,
- decreased adipose tissue expansion,
- improved insulin sensitivity, and
- increased longevity.
Foxa3 gain-of-function and loss-of-function studies in inguinal adipose depots demonstrated
- a cell-autonomous function for Foxa3 in white fat tissue browning.
The mechanisms of Foxa3 modulation of brown fat gene programs involve
- the suppression of peroxisome proliferator activated receptor γ coactivtor 1 α (PGC1α) levels
- through interference with cAMP responsive element binding protein 1-mediated
- transcriptional regulation of the PGC1α promoter.
Our data demonstrate a role for Foxa3 in energy expenditure and in age-associated metabolic disorders.
Control of Mitochondrial pH by Uncoupling Protein 4 in Astrocytes Promotes Neuronal Survival
HP Lambert, M Zenger, G Azarias, Jean-Yves Chatton, PJ. Magistretti,§, S Lengacher
JBC (in press) M114.570879 http://www.jbc.org/cgi/doi/10.1074/jbc.M114.570879
Background: Role of uncoupling proteins (UCP) in the brain is unclear.
Results: UCP, present in astrocytes, mediate the intra-mitochondrial acidification leading to a decrease in mitochondrial ATP production.
Conclusion: Astrocyte pH regulation promotes ATP synthesis by glycolysis whose final product, lactate, increases neuronal survival.
Significance: We describe a new role for a brain uncoupling protein.
Brain activity is energetically costly and requires a steady and
- highly regulated flow of energy equivalents between neural cells.
It is believed that a substantial share of cerebral glucose, the major source of energy of the brain,
- will preferentially be metabolized in astrocytes via aerobic glycolysis.
The aim of this study was to evaluate whether uncoupling proteins (UCPs),
- located in the inner membrane of mitochondria,
- play a role in setting up the metabolic response pattern of astrocytes.
UCPs are believed to mediate the transmembrane transfer of protons
- resulting in the uncoupling of oxidative phosphorylation from ATP production.
UCPs are therefore potentially important regulators of energy fluxes. The main UCP isoforms
- expressed in the brain are UCP2, UCP4, and UCP5.
We examined in particular the role of UCP4 in neuron-astrocyte metabolic coupling
- and measured a range of functional metabolic parameters
- including mitochondrial electrical potential and pH,
- reactive oxygen species production,
- NAD/NADH ratio,
- ATP/ADP ratio,
- CO2 and lactate production, and
- oxygen consumption rate (OCR).
In brief, we found that UCP4 regulates the intra-mitochondrial pH of astrocytes
- which acidifies as a consequence of glutamate uptake,
- with the main consequence of reducing efficiency of mitochondrial ATP production.
- the diminished ATP production is effectively compensated by enhancement of glycolysis.
- this non-oxidative production of energy is not associated with deleterious H2O2 production.
We show that astrocytes expressing more UCP4 produced more lactate,
- used as energy source by neurons, and had the ability to enhance neuronal survival.
Chapter 7: Metabolomics
Introduction to Metabolomics
Author and Curator: Larry H. Bernstein, MD, FCAP
This concludes a long step-by-step journey into rediscovering biological processes from the genome as a framework to the remodeled and reconstituted cell through a number of posttranscription and posttranslation processes that modify the proteome and determine the metabolome. The remodeling process continues over a lifetime. The process requires a balance between nutrient intake, energy utilization for work in the lean body mass, energy reserves, endocrine, paracrine and autocrine mechanisms, and autophagy. It is true when we look at this in its full scope – What a creature is man?
|
|||||||
Metabolomics is the scientific study of chemical processes involving metabolites. Specifically, metabolomics is the “systematic study of the unique chemical fingerprints that specific cellular processes leave behind”, the study of their small-molecule metabolite profiles.[1] The metabolome represents the collection of all metabolites in a biological cell, tissue, organ or organism, which are the end products of cellular processes.[2] mRNA gene expression data and proteomic analyses reveal the set of gene products being produced in the cell, data that represents one aspect of cellular function. Conversely, metabolic profiling can give an instantaneous snapshot of the physiology of that cell. One of the challenges of systems biology andfunctional genomics is to integrate proteomic, transcriptomic, and metabolomic information to provide a better understanding of cellular biology.
The term “metabolic profile” was introduced by Horning, et al. in 1971 after they demonstrated that gas chromatography-mass spectrometry (GC-MS) could be used to measure compounds present in human urine and tissue extracts. The Horning group, along with that of Linus Pauling and Arthur B. Robinson led the development of GC-MS methods to monitor the metabolites present in urine through the 1970s.
Concurrently, NMR spectroscopy, which was discovered in the 1940s, was also undergoing rapid advances. In 1974, Seeley et al. demonstrated the utility of using NMR to detect metabolites in unmodified biological samples.This first study on muscle highlighted the value of NMR in that it was determined that 90% of cellular ATP is complexed with magnesium. As sensitivity has improved with the evolution of higher magnetic field strengths and magic angle spinning, NMR continues to be a leading analytical tool to investigate metabolism. Efforts to utilize NMR for metabolomics have been influenced by the laboratory of Dr. Jeremy Nicholson at Birkbeck College, University of London and later at Imperial College London. In 1984, Nicholson showed 1H NMR spectroscopy could potentially be used to diagnose diabetes mellitus, and later pioneered the application of pattern recognition methods to NMR spectroscopic data.
In 2005, the first metabolomics web database, METLIN, for characterizing human metabolites was developed in the Siuzdak laboratory at The Scripps Research Institute and contained over 10,000 metabolites and tandem mass spectral data. As of September 2012, METLIN contains over 60,000 metabolites as well as the largest repository of tandem mass spectrometry data in metabolomics.
On 23 January 2007, the Human Metabolome Project, led by Dr. David Wishart of the University of Alberta, Canada, completed the first draft of the human metabolome, consisting of a database of approximately 2500 metabolites, 1200 drugs and 3500 food components. Similar projects have been underway in several plant species, most notably Medicago truncatula and Arabidopsis thaliana for several years.
As late as mid-2010, metabolomics was still considered an “emerging field”. Further, it was noted that further progress in the field depended in large part, through addressing otherwise “irresolvable technical challenges”, by technical evolution ofmass spectrometry instrumentation.
Metabolome refers to the complete set of small-molecule metabolites (such as metabolic intermediates, hormones and other signaling molecules, and secondary metabolites) to be found within a biological sample, such as a single organism. The word was coined in analogy with transcriptomics and proteomics; like the transcriptome and the proteome, the metabolome is dynamic, changing from second to second. Although the metabolome can be defined readily enough, it is not currently possible to analyse the entire range of metabolites by a single analytical method. The first metabolite database(called METLIN) for searching m/z values from mass spectrometry data was developed by scientists at The Scripps Research Institute in 2005. In January 2007, scientists at the University of Alberta and theUniversity of Calgary completed the first draft of the human metabolome. They catalogued approximately 2500 metabolites, 1200 drugs and 3500 food components that can be found in the human body, as reported in the literature. This information, available at the Human Metabolome Database (www.hmdb.ca) and based on analysis of information available in the current scientific literature, is far from complete.
Each type of cell and tissue has a unique metabolic ‘fingerprint’ that can elucidate organ or tissue-specific information, while the study of biofluids can give more generalized though less specialized information. Commonly used biofluids are urine and plasma, as they can be obtained non-invasively or relatively non-invasively, respectively. The ease of collection facilitates high temporal resolution, and because they are always at dynamic equilibrium with the body, they can describe the host as a whole.
Metabolites are the intermediates and products of metabolism. Within the context of metabolomics, a metabolite is usually defined as any molecule less than 1 kDa in size.
A primary metabolite is directly involved in the normal growth, development, and reproduction. A secondary metabolite is not directly involved in those processes. By contrast, in human-based metabolomics, it is more common to describe metabolites as being either endogenous (produced by the host organism) orexogenous. Metabolites of foreign substances such as drugs are termed xenometabolites. The metabolome forms a large network of metabolic reactions, where outputs from one enzymatic chemical reaction are inputs to other chemical reactions.
Metabonomics is defined as “the quantitative measurement of the dynamic multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification”. The word origin is from the Greek μεταβολή meaning change and nomos meaning a rule set or set of laws. This approach was pioneered by Jeremy Nicholson at Imperial College London and has been used in toxicology, disease diagnosis and a number of other fields. Historically, the metabonomics approach was one of the first methods to apply the scope of systems biology to studies of metabolism.
There is a growing consensus that ‘metabolomics’ places a greater emphasis on metabolic profiling at a cellular or organ level and is primarily concerned with normal endogenous metabolism. ‘Metabonomics’ extends metabolic profiling to include information about perturbations of metabolism caused by environmental factors (including diet and toxins), disease processes, and the involvement of extragenomic influences, such as gut microflora. This is not a trivial difference; metabolomic studies should, by definition, exclude metabolic contributions from extragenomic sources, because these are external to the system being studied.
Toxicity assessment/toxicology. Metabolic profiling (especially of urine or blood plasma samples) detects the physiological changes caused by toxic insult of a chemical (or mixture of chemicals).
Functional genomics. Metabolomics can be an excellent tool for determining thephenotype caused by a genetic manipulation, such as gene deletion or insertion. Sometimes this can be a sufficient goal in itself—for instance, to detect any phenotypic changes in a genetically-modified plant intended for human or animal consumption. More exciting is the prospect of predicting the function of unknowngenes by comparison with the metabolic perturbations caused by deletion/insertion of known genes.
Nutrigenomics is a generalised term which links genomics, transcriptomics, proteomics and metabolomics to human nutrition. In general a metabolome in a given body fluid is influenced by endogenous factors such as age, sex, body composition and genetics as well as underlying pathologies. The large bowel microflora are also a very significant potential confounder of metabolic profiles and could be classified as either an endogenous or exogenous factor. The main exogenous factors are diet and drugs. Diet can then be broken down to nutrients and non- nutrients.
http://en.wikipedia.org/wiki/Metabolomics
Jose Eduardo des Salles Roselino
The problem with genomics was it was set as explanation for everything. In fact, when something is genetic in nature the genomic reasoning works fine. However, this means whenever an inborn error is found and only in this case the genomic knowledge afterwards may indicate what is wrong and not the completely way to put biology upside down by reading everything in the DNA genetic as well as non-genetic problems.







7.1 Extracellular Evaluation of Intracellular Flux in Yeast Cells
Larry H. Bernstein, MD, FCAP
7.2 Metabolomic Analysis of Two Leukemia Cell Lines Part I
Larry H. Bernstein, MD, FCAP
7.3 Metabolomic Analysis of Two Leukemia Cell Lines Part II
Larry H. Bernstein, MD, FCAP
7.4 Buffering of Genetic Modules involved in Tricarboxylic Acid Cycle Metabolism provides Homeostatic Regulation
Larry H. Bernstein, MD, FCAP
7.5 Metabolomics, Metabonomics and Functional Nutrition: The Next Step in Nutritional Metabolism and Biotherapeutics
Larry H. Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
7.6 Isoenzymes in Cell Metabolic Pathways
Larry H. Bernstein, MD, FCAP
7.7 A Brief Curation of Proteomics, Metabolomics, and Metabolism
Larry H Bernstein, MD, FCAP
7.8 Metabolomics is about Metabolic Systems Integration
Larry H Bernstein, MD, FCAP
7.9 Mechanisms of Drug Resistance
Larry H. Bernstein, MD, FCAP
7.10 Development Of Super-Resolved Fluorescence Microscopy
Larry H Bernstein, MD, FCAP
7.11 Metabolic Reactions Need Just Enough
Larry H Bernstein, MD, FCAP
Summary of Metabolomics
Author and Curator: Larry H. Bernstein, MD, FCAP
This concludes the series on metabolomics, a rapidly developing science that is interconnected with a group termed – OMICS: proteomics, transcriptomics, genomics, and metabolomics. This chapter is most representative of the many important studies being done in the field, which ranges most widely because it has opened doors into nutrition and nutritional supplements, plant biochemistry, agricultural crops and breeding, animal breeding, worldwide malnutrition, diabetes, cancer, neurosciences, circulatory, respiratory, and musculosletal disorders, infectious diseases and immune system disorders. Obviously, it is not possible to cover the full range of activity, but metabolomics is most comprehensive in exploring the full range of metabolic changes that occur in health during the full age range from development to the geriatric years. It can be integrated well with gene expression, proteomics studies, and epidemiological investigations.
The subchapters are given here:
7.1 Extracellular Evaluation of Intracellular Flux in Yeast Cells
7.2 Metabolomic Analysis of Two Leukemia Cell Lines Part I
7.3 Metabolomic Analysis of Two Leukemia Cell Lines Part II
7.6 Isoenzymes in Cell Metabolic Pathways
7.7 A Brief Curation of Proteomics, Metabolomics, and Metabolism
7.8 Metabolomics is about Metabolic Systems Integration
7.9 Mechanisms of Drug Resistance
7.10 Development Of Super-Resolved Fluorescence Microscopy
Metabolomics Summary and Perspective
This chapter will be followed by an exploration of disease and pharmaceutical directed studies using these methods 8. Impairments in pathological states: endocrine disorders, stress hypermetabolism and cancer
Networking metabolites and diseases
P Braun, E Rietman, and M Vidal
Center for Cancer Systems Biology and Department of Cancer Biology, Dana-Farber Cancer Institute, and Department of Genetics, Harvard Medical School, Boston, MA; and Physical Sciences Inc., Andover, MA 01810
PNAS July 22, 2008; 105(29): 9849–9850. http://pnas.org/cgi/doi/10.1073/pnas.0805644105
Biological systems are increasingly viewed and analyzed as
- highly complex networks of interlinked macromolecules and metabolites.
Network analysis has been applied to
- interactome maps of protein–protein, protein–DNA, and protein–RNA interactions
- as well as transcriptional, metabolic, and genetic data.
Such network views of biological systems should facilitate the detection of
- nonlinear long-range effects of perturbations, for example, by mutations, and
- help identification of unanticipated indirect causal connections.
Diseasome and Drug-Target Network
Recently, Goh et al. (1) constructed a ‘‘diseasome’’ network in which
- two diseases are linked to each other if
- they share at least one gene, in which mutations are associated with both diseases.
In the resulting network, related disease families cluster tightly together, thus
- phenotypically defining functional modules.
Importantly, for the first time this study applied concepts from network biology to human diseases,
- thus opening the door for discovering causal relationships between
- disregulated networks and resulting ailments.
Subsequently Yilderim et al. (2) linked drugs to protein targets in a drug–target network,
- which could then be overlaid with the diseasome network.
One notable finding was the recent trend toward the development of
- new compounds directly targeted at disease gene products, whereas previous drugs,
- often found by trial and error, appear to target proteins only indirectly related to
- the actual disease molecular mechanisms.
An important question that remains in this emerging field of network analysis consists of
- investigating the extent to which directly targeting the product of mutated genes is an efficient approach or
- whether targeting network properties instead, and
- thereby accounting for indirect nonlinear effects of system perturbations by drugs, may prove more fruitful.
However, to answer such questions it is important to have a good understanding of the various influences that can lead to diseases.
Metabolic Connections
One group of diseases that was very poorly connected in the original diseasome network was the family of metabolic diseases.
In this issue of PNAS, Lee et al. (3) hypothesize that metabolic diseases may instead be connected
- via metabolites and common reactions.
To investigate this hypothesis Lee et al. first constructed a metabolic network from data available in
- two manually curated databases detailing well known
- metabolic reactions,
- the involved metabolites, and
- catalyzing enzymes.
In addition, gene–disease associations were identified by using the Online Mendelian Inheritance in Man (OMIM) database (http://ncbi.nlm.nih.gov/sites/
entrez?dbomim&itooltoolbar). In a last step,
- a metabolic disease network (MDN) was constructed by connecting
- two diseases if their associated genes are linked in the metabolic network
- by a common metabolite or metabolites used in a common reaction.
Metabolites are not only linked by common reactions, but
- on a larger scale by coupled fluxes within a metabolic network,
- which may also influence disease phenotypes.
An increase in the concentration of one metabolite may increase several fluxes
- across reaction pathways that use this compound, which
- may lead to diverse phenotypes and distinct diseases.
The fluxes within the metabolic network are calculated by using
- the Flux Coupling Finder method described by Nikolaev et al. (4) and Burgard et al. (5),
- which is based on the assumption that pools of metabolites are conserved.
To functionally validate the network, coexpression correlations are measured for genes
- linked by adjacent reactions and those linked by fluxes.
Interestingly, the average coexpression correlation for flux-coupled genes (0.31)
- is higher than that for genes simply catalyzing adjacent reactions (0.24)
(compared with 0.10 for all gene pairs in the network).
If the links between diseases identified in the MDN are functionally and causally relevant
- it should be expected that linked diseases occur more frequently in the same individual.
To test this hypothesis, Lee et al. (3) measured the co-occurrence of diseases in patients by using detailed Medicare information
- of 13 million patients and 32 million hospital visits within a 3-year period.
A comorbidity index was computed to measure the degree to which one disease
- will increase the likelihood of a second disease in the same patient.
The average comorbidity for all genes is 0.0008 (Pearson correlation coefficient),
- which increases 3-fold to 0.0027 when disease pairs that are metabolically linked are analyzed,
- which is highly statistically significant (P 108).
When diseases are analyzed that are directionally coupled by a flux (see ref. 3 for details),
- the correlation increases to 0.0062.
Thus, whereas 17% of all diseases in the network show significant comorbidity, this fraction
- nearly doubles to 31% for metabolically linked diseases.
Further analysis reveals that comorbidity effects can be detected up to three links (metabolites, reactions)
- apart from each other with statistical significance, but not farther away.
In the MDN, several highly connected hubs, e.g., hypertension and hemolytic anemia, are
- linked to many different co-occurring diseases not unexpected for such complex diseases
- that can result from many different genetic alterations or variants.
Importantly, though, most of the connections to the different linked diseases
- are mediated by diverse connections in the metabolic network.
Thus, in the future such insights may be helpful for finer classification of the complex hub disease.
Furthermore, depending on the onset of the complex (hub) disease in relation to the associated diseases,
- such relationships may potentially be used to systematically
- stratify patients and develop targeted treatments acting on
- the underlying metabolic links.
Returning to the starting point of their study, Lee et al. (3) next investigated
- whether metabolic diseases are better linked through the metabolic network
- than they are in the previously described gene–disease network.
When purely metabolic diseases are considered, the comorbidity is, in fact,
- best predicted by metabolic links.
Interestingly, when all diseases linked to metabolic enzymes are considered,
- which involves many diseases that are merely related to metabolic diseases through multifunctional enzymes,
- the gene and metabolic networks are nearly equally predictive of comorbidity,
- indicating that as a general approach information from
- many different biological dimensions should be integrated to identify the most relevant connections.
Together, all these findings support the initial hypothesis that metabolic diseases are linked by metabolic networks.
Practically, alteration of one metabolite or one reaction can have numerous repercussions in the network,
- each of which can manifest as different diseases that frequently occur together in affected patients.
Radoslav Bozov
- Glycine, as the only amino acid having no isomer driven central carbon allowing for hing occupancy of ‘free’
motifs, where quark (proton) ‘fluxes’ play at, is a one – step away observable (1) from synthesis of pyrimidines
to glyoxylate mitochondrial ‘shunt’ entangling at least two differential compartments longly objected by
Japanese metabolomics study groups. - One carbon systems emerge out of a glycoprotein ‘complex’, pyrimidine synthase pathway, that possesses
significant similarity to BRCA2 and most other transcription factors suggesting that protein allocation is
coorchestrated by modifications and spatially transforming construes as an outcome of energy processing.
Directly deduced by TCS, life cannot exist without mutations, as mutations and chromatin states appear to
be a sort of energy hold and release ‘gates’. - Phosphorylations and small molecules as such as cGMP, cAMP play a role of decompression machinery
for amplifying bio signal processing C-S, C-N, C-O, interference open systems. By decompression
of one relative discrete space, another one becomes compressed, which gets uncertainty of absolute energy
processing within space scalar wise into vector objected space represented by chromatin remodeling processes,
possibly seen as network identities information. - Unifying network and quantum theory possess implications to relativity concepts and energy relevant computational methodology.






Chapter 8. Impairments in Pathological States: Endocrine Disorders; Stress Hypermetabolism and Cancer
Introduction to Impairments in Pathological States: Endocrine Disorders, Stress Hypermetabolism and Cancer
Author and Curator: Larry H. Bernstein, MD, FCAP
This leads into a series of presentations and the metabolic imbalance central to findings of endocrine, metabolic, inflammatory, immune diseases and cancer. All of this has been a result of discoveries based on the methods of study of genomiocs, proteomics, transcriptomics, and metabolomics that have preceded this. In some cases there has been the use of knockout methods. The completion of the human genomic and other catalogues have been instrumental in the past few years. In all cases there has been a thorough guidance by a biological concept of mechanism based on gene expression, metabolic disturbance, signaling pathways, and up- or down- regulation of metabolic circuits. It is interesting to recall that a concept of metabolic circuits was not yet formulated at the time of the mid 20th century physiology, except perhaps with respect to the coagulation pathways, and to some extent, glycolysis, gluconeogenesis, the hexose monophosphate shunt, and mitochondrial respiration, which were linear strings of enzyme substrate reactions that intersected and that had flow restraints not then understood as to the complexity we now appreciate. We did know the importance of cytochrome c, the adenine and pyridine nucleotides, and the energy balance. Electron microscopy had opened the door to understanding the mechanism of contraction of skeletal muscle and myocardium, but it also opened the door to understanding kidney structure and function, explaining the “mesangium”. The first cardiac maker was discovered by Arthur Karmen in the serum alanine and aspartate aminotransferases, with a consequent differentiation between hepatic and myocardial damage. This was followed by lactic dehydrogenase and the H- and M-type isoenzymes in the 1960s, and in the next decade, by the MB-isoenzyme of creatine kinase. Troponins T and then I would not be introduced until the mid 1980s, and they have become a gold standard for the diagnosis of myocardial infarction.
In the 1980s we also saw the development of antiplatelet therapy that rapidly advanced interventional cardiology. But advances in surgical as well as medical intervention also proceeded as the understanding of the lipid metabolism was opened by the work of Brown and Goldstein, and UTSW Medical Campus, and major advances in treatment came at Baylor and UT Medical Center in Houston, and at the Cleveland Clinic. The next important advance came with the discovery of nitric oxide synthase role in endothelium and oxidative stress. The field of endocrinology saw advances as well for a solid period of 30 years in a comparable period for the adrenals, thyroid, and pituitary glands, and for the understanding of the male and female sex hormones, and discoveries in breast, ovarian, and prostate cancer. There were cancer markers, such as, CA125 and CA15-3, and PSA. This had more of an impact on timely surgical intervention, and if not that, post surgical followup. Despite a long time into the war on cancer, introduced by President Lynden Johnson, the fundamental knowledge needed was not sufficient. In the meantime, there were advances in the treatment of diabetes, with eventual introduction of the insulin pump for type I diabetes. The problem of Type 2 DM increased in prevalence, reaching into the childhood age group, with ascendent obesity. An epidemiological pattern of disease comorbidities was emergent. Our population has aged out, and with it we are seeing an increase in dementias, especially Alzheimer’s disease. But the knowledge of the brain has lagged far behind.
What follows is a series of chapters that address what has currently been advanced with repect to the alignment of our knowledge of the last decade and pharmacetical discovery. Pharmaceuticals were suitable for bacterial infections until the 1990s, when we saw the rise of resistance to penicillins and Vancomycin, and we had issues with gram negative enterobacter, salmonella, and E. coli strains. That has been and is a significant challenge. The elucidation of the gut microbiome in recent years will help to relieve this problem. The problem of the variety and different aggressive types of cancer has been another challenge. The door has been opened to better diagnostic tools with respsct to imaging and targeted biomarkers for localization. I am not dealing with imaging, which is not the subject here.
HLA targeting efficiency correlates with human T-cell response magnitude and with mortality from influenza A infection
From http://www.pnas.org – Sep 3, 2013 4:24 PM
Experimental and computational evidence suggests that HLAs preferentially bind
- conserved regions of viral proteins, a concept we term “targeting efficiency,” and
- that this preference may provide improved clearance of infection in several viral systems.
To test this hypothesis, T-cell responses to A/H1N1 (2009)
- were measured from peripheral blood mononuclear cells
- obtained from a household cohort study performed during the 2009–2010 influenza season.
We found that HLA targeting efficiency scores
- significantly correlated with IFN-γ
enzyme-linked immunosorbent spot responses (P = 0.042, multiple regression).
A further population-based analysis found that
- the carriage frequencies of the alleles with the lowest targeting efficiencies, A*24,
- were associated with pH1N1 mortality (r = 0.37, P = 0.031) and
- are common in certain indigenous populations in which
- increased pH1N1 morbidity has been reported.
HLA efficiency scores and HLA use are associated with
- CD8 T-cell magnitude in humans after influenza infection.
The computational tools used in this study may be useful predictors of
- potential morbidity and identify immunologic differences of new variant influenza strains
- more accurately than evolutionary sequence comparisons.
Population-based studies of the relative frequency of these alleles
- in severe vs. mild influenza cases might advance clinical practices
- for severe H1N1 infections among genetically susceptible populations.
A deeper look into cholesterol synthesis
By Swathi Parasuraman
The human body needs cholesterol to maintain membrane fluidity, and
- it acts as a precursor molecule for several important biochemical pathways.
Its regulation requires strict control, as it can cause problems if it’s produced in excess. In 1964, Konrad Bloch received a Nobel Prize for his work elucidating the mechanisms of cholesterol synthesis. His work
- eventually contributed to the discovery of statins, drugs used today to lower blood cholesterol levels.
The biosynthesis of cholesterol is a complex process with more than 20 steps. One of the first enzymes is
- 3-hydroxy-3-methylglutaryl-CoA reductase, also known as HMGCR, the main target of statins.
As links between intermediates in cholesterol synthesis and various diseases are being discovered continually, more information about the regulatory role of the post-HMGCR pathway is needed.
In a recent minireview in The Journal of Biological Chemistry, Laura Sharpe and Andrew Brown of the University of New South Wales describe
- multiple ways various enzymes other than HMGCR
- are implicated in the modulation of cholesterol synthesis.
One such enzyme is squalene monooxygenase, which, like HMGCR, can be destroyed
- by the proteasome when cholesterol levels are high.
The minireview also explains how pathway intermediates
- can have functions distinct from those of cholesterol.
For example, intermediate 7-dehydrocholesterol usually is converted to cholesterol by the enzyme DHCR7
- but is also a vitamin D precursor.
To synthesize the enzymes necessary to make cholesterol,
- SREBPs, short for sterol regulatory element binding proteins, have special functions.
Along with transcriptional cofactors, they activate gene expression
- in response to low sterol levels and, conversely,
- are suppressed when there is enough cholesterol around.
Additionally, SREBPs control production of
- nicotinamide adenine dinucleotide phosphate, or NADPH,
- which is the reducing agent required to carry out the different steps in the pathway.
Lipid carrier proteins also can facilitate cholesterol synthesis. One example is SPF, or supernatant protein factor,
- which transfers substrate from an inactive to an active pool or
- from one enzyme site to another.
Furthermore, translocation of several cholesterogenic enzymes
- from the endoplasmic reticulum to other cell compartments can occur under various conditions,
- thereby regulating levels and sites of intracellular cholesterol accumulation.
Immunology in the gut mucosa:
20 Feb 2013 by Kausik Datta, posted in Immunology, Science (Nature)
The human gut can be the scene for devastating conditions such as inflammatory bowel disease,
- which arises through an improperly controlled immune response.
The gut is often the body’s first point of contact with microbes; every mouthful of food is accompanied by a cargo of micro-organisms that go on to encounter the mucosa, the innermost layer of the gut. Most microbes are destroyed by the harsh acidic environment in the stomach, but a hardy few make it through to the intestines.
The intestinal surface is covered with finger-like protrusions called villi,
whose primary function is the absorption of nutrients.
These structures and the underlying tissues
- host the body’s largest population of immune cells.
Scattered along the intestinal mucosa are
- dome-like structures called Peyer’s Patches.
These are enriched in lymphoid tissue, making them key sites for
- coordinating immune responses to pathogens,
- whilst promoting tolerance to harmless microbes and food.
The villi contain a network of blood vessels to transport nutrients from food to the rest of the body. Lymphatics
- from both the Peyer’s Patches and the villi
- drain into the mesenteric lymph nodes.
Within the villi is a network of loose connective tissue called the lamina propria, and
- at the base of the villi are the crypts which host the stem cells that replenish the epithelium.
The epithelium together with its overlying mucus forms
- a barrier against microbial invasion.
A mix of immune cells including T- and B-lymphocytes, macrophages, and dendritic cells are
- embedded within the matrix of the Peyer’s Patches, .
A key function of the Peyer’s Patch is the sampling of antigens present in the gut. The Peyer’s Patch has a thin mucous layer and specialized phagocytic cells, called M-cells, which
- transport material across the epithelial barrier via a process called transcytosis.
Dendritic cells extend dendrites between epithelial cells to sample antigens that are then
- broken down and used for presenting to lymphocytes.
Sampling antigens in this way typically results in so-called tolerogenic activation, where
- the immune system initiates an anti-inflammatory response.
With their cargo of antigens, these Dendritic Cells then
- traffic to the T-cell zones of the Peyer’s Patch.
Upon encounter with specific T-cells, the Dendritic Cells
- convert them into an immunomodulatory cell called regulatory T-cell or T-reg.
Defects in the function of these cells are associated with
- inflammatory bowel disease in both animals and humans.
These T-regs migrate to lamina propria of the villi via the lymphatics. Here, the T-regs
- secrete a molecule called Interleukin (IL)-10,
- which exerts a suppressive action on immune cells within the lamina propria
- and upon the epithelial layer itself.
IL10 is, therefore, critical in maintaining immune quiescence
- and preventing unnecessary inflammation.
However, a breakdown in this process of immune homeostasis results in gut pathology and
- when this occurs over a prolonged period and in an uncontrolled manner,
- it can lead to inflammatory bowel disease.
Chemical, mechanical or pathogen-triggered barrier disruption
- coupled with particular genetic susceptibilities may all combine to set off inflammation.
Epithelium coming into contact with bacteria
- is activated, leading to bacterial influx.
Alarm molecules released by the epithelium
- activates immune cells, and T-regs in the vicinity
- scale down their IL10 secretion to enable an immune response to proceed.
Dendritic cells are also activated by this environment, and
- start to release key inflammatory molecules,
- such as IL6, IL12, and IL23.
Effector T-cells also appear on the scene and
- these coordinate an escalation of the immune response
- by secreting their own inflammatory molecules,
- Tumor Necrosis Factor (TNF)-α, Interferon (IFN)-γ and IL17.
Soon after the effector T-cells are arrived, a voracious phagocyte called a neutrophil is recruited. Neutrophils are critical for the clearance of the bacteria. One weapon in the neutrophil armory is
- the ability to undergo self-destruction.
This leaves behind a jumble of DNA saturated with enzymes, called the Neutrophil Extracellular Trap.
Although this can effectively destroy the bacterial invaders
- and plug any breaches in the epithelial wall,
- it also causes collateral damage to tissues.
Slowly the tide begins to turn and the bacterial invasion is repulsed. Any remaining neutrophils die off,
- and are cleared by macrophages.
Epithelial integrity is restored by replacement of damaged cells with new ones from the intestinal crypts. Finally T-regs are recruited once again to calm the immune response.
Targeting the molecules involved in gut pathology is leading to
- effective therapies for inflammatory bowel disease.
Notes:
T- and B-lymphocytes, Macrophages, and Dendritic Cells: These are all important immune effector cells. Macrophages and Dendritic cells are primary defence cells that can eat up (‘phagocytosis’) microbes and destroy them; they also can present parts of these microbes to lymphocytes. T-lymphocytes or T-cells help B-lymphocytes or B-cells recognize the antigen and form antibodies against it. Other types of T-cells can themselves kill microbes. All these cells also secrete various chemical substances, called cytokines and chemokines, which act as molecular messengers in recruiting various immune cells, coordinating and fine-tuning the immune response. Some of these cytokines are called Interleukins, shortened to IL.
Anti-inflammatory response: A type of immune response in which molecular messengers are used to scale down heavy-handed immune cell activity and switch off processes that recruit immune cells. This helps the body recognize and selectively tolerate beneficial substances such as commensalic microbes that live in the gut.
Neutrophils: These are highly versatile immune effector cells. Usually, they are one of the first cells recruited to the site of infection or tissue damage via message spread by molecular messengers. Neutrophils can themselves elaborate cytokines and chemokines, and have the ability to directly kill microbes.
Oxazoloisoindolinones with in vitro antitumor activity selectively activate a p53-pathway through potential inhibition of the p53-MDM2 interaction.
J Soares, et al. Eur J Pharm Sci 10/2014;http://dx.doi.org:/10.1016/j.ejps.2014.10.006
An appealing target for anticancer treatment is
- the p53 tumor suppressor protein.
This protein is inactivated in half of human tumors
- due to endogenous negative regulators such as MDM2.
Therefore, restoring the p53 activity through
- the inhibition of its interaction with MDM2
- is considered a valuable therapeutic strategy
- against cancers with a wild-type p53 status.
We report the synthesis of nine enantiopure phenylalaninol-derived oxazolopyrrolidone lactams
- and the evaluation of their biological effects as p53-MDM2 interaction inhibitors.
Using a yeast-based screening assay, two oxazoloisoindolinones,
- were identified as potential p53-MDM2 inhibitors.
The molecular mechanism of oxazoloisoindolinone 3a validated
- in human colon adenocarcinoma HCT116 cells with wild-type p53 (HCT116 p53(+/+)) and
- in its isogenic derivative without p53 (HCT116 p53(-/-)).
we demonstrated that oxazoloisoindolinone 3a exhibited
- a p53-dependent in vitro antitumor activity through
- induction of G0/G1-phase cell cycle arrest and apoptosis.
The selective activation of a p53-apoptotic pathway by oxazoloisoindolinone 3a was further supported
- by the occurrence of PARP cleavage only in p53-expressing HCT116 cells.
Oxazoloisoindolinone 3a led
- to p53 protein stabilization
- to the up-regulation of p53 transcriptional activity &
- increased expression levels of several p53 target genes,
- as p21, MDM2, BAX and PUMA,
- in p53(+/+) but not in p53(-/-) HCT116 cells.
the ability of oxazoloisoindolinone 3a to block the p53-MDM2 interaction in HCT116 p53(+/+) cells was confirmed by co-immunoprecipitation.
molecular docking analysis of the interactions
- between the compounds and MDM2 revealed that
- oxazoloisoindolinone 3a binds to MDM2.
this work adds the oxazoloisoindolinone scaffold to the activators of a wild-typep53-pathway with promising antitumor activity.
it may open the way to the development of
- a new class of p53-MDM2 interaction inhibitors.
TrypanoCyc: a community-led biochemical pathways database for Trypanosoma brucei.
Sanu Shameer, et al. Nucleic Acids Research10/2014;
http://dx.doi.org/10.1093/nar/gku944
The metabolic network of a cell represents the catabolic and anabolic reactions that interconvert small molecules (metabolites) through the activity of enzymes, transporters and non-catalyzed chemical reactions. Our understanding of individual metabolic networks is increasing as we learn more about the enzymes that are active in particular cells under particular conditions and as technologies advance to allow detailed measurements of the cellular metabolome.
Metabolic network databases are important in allowing us to
- contextualise data sets emerging from transcriptomic, proteomic and metabolomic experiments.
Here we present a dynamic database, TrypanoCyc (http://www.metexplore.fr/trypanocyc/), which describes
- the generic and condition-specific metabolic network of Trypanosoma brucei, a parasitic protozoan
- responsible for human and animal African trypanosomiasis.
In addition to enabling navigation through the BioCyc-based TrypanoCyc interface, we have implemented a network
- representation of the information through MetExplore,
yielding a novel environment in which to visualise the metabolism of this important parasite.
Summary and Perspectives: Impairments in Pathological States: Endocrine Disorders, Stress Hypermetabolism and Cancer
Author and Curator: Larry H. Bernstein, MD, FCAP
This summary is the last of a series on the impact of transcriptomics, proteomics, and metabolomics on disease investigation, and the sorting and integration of genomic signatures and metabolic signatures to explain phenotypic relationships in variability and individuality of response to disease expression and how this leads to pharmaceutical discovery and personalized medicine. We have unquestionably better tools at our disposal than has ever existed in the history of mankind, and an enormous knowledge-base that has to be accessed. I shall conclude here these discussions with the powerful contribution to and current knowledge pertaining to biochemistry, metabolism, protein-interactions, signaling, and the application of the -OMICS to diseases and drug discovery at this time.
The Ever-Transcendent Cell
Deriving physiologic first principles By John S. Torday | The Scientist Nov 1, 2014
http://www.the-scientist.com/?articles.view/articleNo/41282/title/The-Ever-Transcendent-Cell/
Both the developmental and phylogenetic histories of an organism describe the evolution of physiology—the complex of metabolic pathways that govern the function of an organism as a whole. The necessity of establishing and maintaining homeostatic mechanisms began at the cellular level, with the very first cells, and homeostasis provides the underlying selection pressure fueling evolution.
While the events leading to the formation of the first functioning cell are debatable, a critical one was certainly the formation of simple lipid-enclosed vesicles, which provided a protected space for the evolution of metabolic pathways. Protocells evolved from a common ancestor that experienced environmental stresses early in the history of cellular development, such as acidic ocean conditions and low atmospheric oxygen levels, which shaped the evolution of metabolism.
The reduction of evolution to cell biology may answer the perennially unresolved question of why organisms return to their unicellular origins during the life cycle.
As primitive protocells evolved to form prokaryotes and, much later, eukaryotes, changes to the cell membrane occurred that were critical to the maintenance of chemiosmosis, the generation of bioenergy through the partitioning of ions. The incorporation of cholesterol into the plasma membrane surrounding primitive eukaryotic cells marked the beginning of their differentiation from prokaryotes. Cholesterol imparted more fluidity to eukaryotic cell membranes, enhancing functionality by increasing motility and endocytosis. Membrane deformability also allowed for increased gas exchange.
Acidification of the oceans by atmospheric carbon dioxide generated high intracellular calcium ion concentrations in primitive aquatic eukaryotes, which had to be lowered to prevent toxic effects, namely the aggregation of nucleotides, proteins, and lipids. The early cells achieved this by the evolution of calcium channels composed of cholesterol embedded within the cell’s plasma membrane, and of internal membranes, such as that of the endoplasmic reticulum, peroxisomes, and other cytoplasmic organelles, which hosted intracellular chemiosmosis and helped regulate calcium.
As eukaryotes thrived, they experienced increasingly competitive pressure for metabolic efficiency. Engulfed bacteria, assimilated as mitochondria, provided more bioenergy. As the evolution of eukaryotic organisms progressed, metabolic cooperation evolved, perhaps to enable competition with biofilm-forming, quorum-sensing prokaryotes. The subsequent appearance of multicellular eukaryotes expressing cellular growth factors and their respective receptors facilitated cell-cell signaling, forming the basis for an explosion of multicellular eukaryote evolution, culminating in the metazoans.
Casting a cellular perspective on evolution highlights the integration of genotype and phenotype. Starting from the protocell membrane, the functional homolog for all complex metazoan organs, it offers a way of experimentally determining the role of genes that fostered evolution based on the ontogeny and phylogeny of cellular processes that can be traced back, in some cases, to our last universal common ancestor. ….
As eukaryotes thrived, they experienced increasingly competitive pressure for metabolic efficiency. Engulfed bacteria, assimilated as mitochondria, provided more bioenergy. As the evolution of eukaryotic organisms progressed, metabolic cooperation evolved, perhaps to enable competition with biofilm-forming, quorum-sensing prokaryotes. The subsequent appearance of multicellular eukaryotes expressing cellular growth factors and their respective receptors facilitated cell-cell signaling, forming the basis for an explosion of multicellular eukaryote evolution, culminating in the metazoans.
Casting a cellular perspective on evolution highlights the integration of genotype and phenotype. Starting from the protocell membrane, the functional homolog for all complex metazoan organs, it offers a way of experimentally determining the role of genes that fostered evolution based on the ontogeny and phylogeny of cellular processes that can be traced back, in some cases, to our last universal common ancestor.
Given that the unicellular toolkit is complete with all the traits necessary for forming multicellular organisms (Science, 301:361-63, 2003), it is distinctly possible that metazoans are merely permutations of the unicellular body plan. That scenario would clarify a lot of puzzling biology: molecular commonalities between the skin, lung, gut, and brain that affect physiology and pathophysiology exist because the cell membranes of unicellular organisms perform the equivalents of these tissue functions, and the existence of pleiotropy—one gene affecting many phenotypes—may be a consequence of the common unicellular source for all complex biologic traits. …
The cell-molecular homeostatic model for evolution and stability addresses how the external environment generates homeostasis developmentally at the cellular level. It also determines homeostatic set points in adaptation to the environment through specific effectors, such as growth factors and their receptors, second messengers, inflammatory mediators, crossover mutations, and gene duplications. This is a highly mechanistic, heritable, plastic process that lends itself to understanding evolution at the cellular, tissue, organ, system, and population levels, mediated by physiologically linked mechanisms throughout, without having to invoke random, chance mechanisms to bridge different scales of evolutionary change. In other words, it is an integrated mechanism that can often be traced all the way back to its unicellular origins.
The switch from swim bladder to lung as vertebrates moved from water to land is proof of principle that stress-induced evolution in metazoans can be understood from changes at the cellular level.
http://www.the-scientist.com/Nov2014/TE_21.jpg
{kind=link}
A MECHANISTIC BASIS FOR LUNG DEVELOPMENT: Stress from periodic atmospheric hypoxia (1) during vertebrate adaptation to land enhances positive selection of the stretch-regulated parathyroid hormone-related protein (PTHrP) in the pituitary and adrenal glands. In the pituitary (2), PTHrP signaling upregulates the release of adrenocorticotropic hormone (ACTH) (3), which stimulates the release of glucocorticoids (GC) by the adrenal gland (4). In the adrenal gland, PTHrP signaling also stimulates glucocorticoid production of adrenaline (5), which in turn affects the secretion of lung surfactant, the distension of alveoli, and the perfusion of alveolar capillaries (6). PTHrP signaling integrates the inflation and deflation of the alveoli with surfactant production and capillary perfusion. THE SCIENTIST STAFF
From a cell-cell signaling perspective, two critical duplications in genes coding for cell-surface receptors occurred during this period of water-to-land transition—in the stretch-regulated parathyroid hormone-related protein (PTHrP) receptor gene and the β adrenergic (βA) receptor gene. These gene duplications can be disassembled by following their effects on vertebrate physiology backwards over phylogeny. PTHrP signaling is necessary for traits specifically relevant to land adaptation: calcification of bone, skin barrier formation, and the inflation and distention of lung alveoli. Microvascular shear stress in PTHrP-expressing organs such as bone, skin, kidney, and lung would have favored duplication of the PTHrP receptor, since sheer stress generates radical oxygen species (ROS) known to have this effect and PTHrP is a potent vasodilator, acting as an epistatic balancing selection for this constraint.
Positive selection for PTHrP signaling also evolved in the pituitary and adrenal cortex (see figure on this page), stimulating the secretion of ACTH and corticoids, respectively, in response to the stress of land adaptation. This cascade amplified adrenaline production by the adrenal medulla, since corticoids passing through it enzymatically stimulate adrenaline synthesis. Positive selection for this functional trait may have resulted from hypoxic stress that arose during global episodes of atmospheric hypoxia over geologic time. Since hypoxia is the most potent physiologic stressor, such transient oxygen deficiencies would have been acutely alleviated by increasing adrenaline levels, which would have stimulated alveolar surfactant production, increasing gas exchange by facilitating the distension of the alveoli. Over time, increased alveolar distension would have generated more alveoli by stimulating PTHrP secretion, impelling evolution of the alveolar bed of the lung.
This scenario similarly explains βA receptor gene duplication, since increased density of the βA receptor within the alveolar walls was necessary for relieving another constraint during the evolution of the lung in adaptation to land: the bottleneck created by the existence of a common mechanism for blood pressure control in both the lung alveoli and the systemic blood pressure. The pulmonary vasculature was constrained by its ability to withstand the swings in pressure caused by the systemic perfusion necessary to sustain all the other vital organs. PTHrP is a potent vasodilator, subserving the blood pressure constraint, but eventually the βA receptors evolved to coordinate blood pressure in both the lung and the periphery.
Gut Microbiome Heritability
Analyzing data from a large twin study, researchers have homed in on how host genetics can shape the gut microbiome.
By Tracy Vence | The Scientist Nov 6, 2014
Previous research suggested host genetic variation can influence microbial phenotype, but an analysis of data from a large twin study published in Cell today (November 6) solidifies the connection between human genotype and the composition of the gut microbiome. Studying more than 1,000 fecal samples from 416 monozygotic and dizygotic twin pairs, Cornell University’s Ruth Ley and her colleagues have homed in on one bacterial taxon, the family Christensenellaceae, as the most highly heritable group of microbes in the human gut. The researchers also found that Christensenellaceae—which was first described just two years ago—is central to a network of co-occurring heritable microbes that is associated with lean body mass index (BMI). …
Of particular interest was the family Christensenellaceae, which was the most heritable taxon among those identified in the team’s analysis of fecal samples obtained from the TwinsUK study population.
While microbiologists had previously detected 16S rRNA sequences belonging to Christensenellaceae in the human microbiome, the family wasn’t named until 2012. “People hadn’t looked into it, partly because it didn’t have a name . . . it sort of flew under the radar,” said Ley.
Ley and her colleagues discovered that Christensenellaceae appears to be the hub in a network of co-occurring heritable taxa, which—among TwinsUK participants—was associated with low BMI. The researchers also found that Christensenellaceae had been found at greater abundance in low-BMI twins in older studies.
To interrogate the effects of Christensenellaceae on host metabolic phenotype, the Ley’s team introduced lean and obese human fecal samples into germ-free mice. They found animals that received lean fecal samples containing more Christensenellaceae showed reduced weight gain compared with their counterparts. And treatment of mice that had obesity-associated microbiomes with one member of the Christensenellaceae family, Christensenella minuta, led to reduced weight gain. …
Ley and her colleagues are now focusing on the host alleles underlying the heritability of the gut microbiome. “We’re running a genome-wide association analysis to try to find genes—particular variants of genes—that might associate with higher levels of these highly heritable microbiota. . . . Hopefully that will point us to possible reasons they’re heritable,” she said. “The genes will guide us toward understanding how these relationships are maintained between host genotype and microbiome composition.”
J.K. Goodrich et al., “Human genetics shape the gut microbiome,” Cell, http://dx.doi.org:/10.1016/j.cell.2014.09.053, 2014.
Light-Operated Drugs
Scientists create a photosensitive pharmaceutical to target a glutamate receptor.
By Ruth Williams | The Scentist Nov 1, 2014
http://www.the-scientist.com/?articles.view/articleNo/41279/title/Light-Operated-Drugs/

http://www.the-scientist.com/Nov2014/MO1.jpg
{kind=link}
The desire for temporal and spatial control of medications to minimize side effects and maximize benefits has inspired the development of light-controllable drugs, or optopharmacology. Early versions of such drugs have manipulated ion channels or protein-protein interactions, “but never, to my knowledge, G protein–coupled receptors [GPCRs], which are one of the most important pharmacological targets,” says Pau Gorostiza of the Institute for Bioengineering of Catalonia, in Barcelona.
Gorostiza has taken the first step toward filling that gap, creating a photosensitive inhibitor of the metabotropic glutamate 5 (mGlu5) receptor—a GPCR expressed in neurons and implicated in a number of neurological and psychiatric disorders. The new mGlu5 inhibitor—called alloswitch-1—is based on a known mGlu receptor inhibitor, but the simple addition of a light-responsive appendage, as had been done for other photosensitive drugs, wasn’t an option. The binding site on mGlu5 is “extremely tight,” explains Gorostiza, and would not accommodate a differently shaped molecule. Instead, alloswitch-1 has an intrinsic light-responsive element.
In a human cell line, the drug was active under dim light conditions, switched off by exposure to violet light, and switched back on by green light. When Gorostiza’s team administered alloswitch-1 to tadpoles, switching between violet and green light made the animals stop and start swimming, respectively.
The fact that alloswitch-1 is constitutively active and switched off by light is not ideal, says Gorostiza. “If you are thinking of therapy, then in principle you would prefer the opposite,” an “on” switch. Indeed, tweaks are required before alloswitch-1 could be a useful drug or research tool, says Stefan Herlitze, who studies ion channels at Ruhr-Universität Bochum in Germany. But, he adds, “as a proof of principle it is great.” (Nat Chem Biol, http://dx.doi.org:/10.1038/nchembio.1612, 2014)
Enhanced Enhancers
The recent discovery of super-enhancers may offer new drug targets for a range of diseases.
By Eric Olson | The Scientist Nov 1, 2014
http://www.the-scientist.com/?articles.view/articleNo/41281/title/Enhanced-Enhancers/
To understand disease processes, scientists often focus on unraveling how gene expression in disease-associated cells is altered. Increases or decreases in transcription—as dictated by a regulatory stretch of DNA called an enhancer, which serves as a binding site for transcription factors and associated proteins—can produce an aberrant composition of proteins, metabolites, and signaling molecules that drives pathologic states. Identifying the root causes of these changes may lead to new therapeutic approaches for many different diseases.
Although few therapies for human diseases aim to alter gene expression, the outstanding examples—including antiestrogens for hormone-positive breast cancer, antiandrogens for prostate cancer, and PPAR-γ agonists for type 2 diabetes—demonstrate the benefits that can be achieved through targeting gene-control mechanisms. Now, thanks to recent papers from laboratories at MIT, Harvard, and the National Institutes of Health, researchers have a new, much bigger transcriptional target: large DNA regions known as super-enhancers or stretch-enhancers. Already, work on super-enhancers is providing insights into how gene-expression programs are established and maintained, and how they may go awry in disease. Such research promises to open new avenues for discovering medicines for diseases where novel approaches are sorely needed.
Super-enhancers cover stretches of DNA that are 10- to 100-fold longer and about 10-fold less abundant in the genome than typical enhancer regions (Cell, 153:307-19, 2013). They also appear to bind a large percentage of the transcriptional machinery compared to typical enhancers, allowing them to better establish and enforce cell-type specific transcriptional programs (Cell, 153:320-34, 2013).
Super-enhancers are closely associated with genes that dictate cell identity, including those for cell-type–specific master regulatory transcription factors. This observation led to the intriguing hypothesis that cells with a pathologic identity, such as cancer cells, have an altered gene expression program driven by the loss, gain, or altered function of super-enhancers.
Sure enough, by mapping the genome-wide location of super-enhancers in several cancer cell lines and from patients’ tumor cells, we and others have demonstrated that genes located near super-enhancers are involved in processes that underlie tumorigenesis, such as cell proliferation, signaling, and apoptosis.
Super-enhancers cover stretches of DNA that are 10- to 100-fold longer and about 10-fold less abundant in the genome than typical enhancer regions.
Genome-wide association studies (GWAS) have found that disease- and trait-associated genetic variants often occur in greater numbers in super-enhancers (compared to typical enhancers) in cell types involved in the disease or trait of interest (Cell, 155:934-47, 2013). For example, an enrichment of fasting glucose–associated single nucleotide polymorphisms (SNPs) was found in the stretch-enhancers of pancreatic islet cells (PNAS, 110:17921-26, 2013). Given that some 90 percent of reported disease-associated SNPs are located in noncoding regions, super-enhancer maps may be extremely valuable in assigning functional significance to GWAS variants and identifying target pathways.
Because only 1 to 2 percent of active genes are physically linked to a super-enhancer, mapping the locations of super-enhancers can be used to pinpoint the small number of genes that may drive the biology of that cell. Differential super-enhancer maps that compare normal cells to diseased cells can be used to unravel the gene-control circuitry and identify new molecular targets, in much the same way that somatic mutations in tumor cells can point to oncogenic drivers in cancer. This approach is especially attractive in diseases for which an incomplete understanding of the pathogenic mechanisms has been a barrier to discovering effective new therapies.
Another therapeutic approach could be to disrupt the formation or function of super-enhancers by interfering with their associated protein components. This strategy could make it possible to downregulate multiple disease-associated genes through a single molecular intervention. A group of Boston-area researchers recently published support for this concept when they described inhibited expression of cancer-specific genes, leading to a decrease in cancer cell growth, by using a small molecule inhibitor to knock down a super-enhancer component called BRD4 (Cancer Cell, 24:777-90, 2013). More recently, another group showed that expression of the RUNX1 transcription factor, involved in a form of T-cell leukemia, can be diminished by treating cells with an inhibitor of a transcriptional kinase that is present at the RUNX1 super-enhancer (Nature, 511:616-20, 2014).
Fungal effector Ecp6 outcompetes host immune receptor for chitin binding through intrachain LysM dimerization
Andrea Sánchez-Vallet, et al. eLife 2013;2:e00790 http://elifesciences.org/content/2/e00790#sthash.LnqVMJ9p.dpuf

http://img.scoop.it/ZniCRKQSvJOG18fHbb4p0Tl72eJkfbmt4t8yenImKBVvK0kTmF0xjctABnaLJIm9
While host immune receptors
- detect pathogen-associated molecular patterns to activate immunity,
- pathogens attempt to deregulate host immunity through secreted effectors.
Fungi employ LysM effectors to prevent
- recognition of cell wall-derived chitin by host immune receptors
Structural analysis of the LysM effector Ecp6 of
- the fungal tomato pathogen Cladosporium fulvum reveals
- a novel mechanism for chitin binding,
- mediated by intrachain LysM dimerization,
leading to a chitin-binding groove that is deeply buried in the effector protein.
This composite binding site involves
- two of the three LysMs of Ecp6 and
- mediates chitin binding with ultra-high (pM) affinity.
The remaining singular LysM domain of Ecp6 binds chitin with
- low micromolar affinity but can nevertheless still perturb chitin-triggered immunity.
Conceivably, the perturbation by this LysM domain is not established through chitin sequestration but possibly through interference with the host immune receptor complex.
Mutated Genes in Schizophrenia Map to Brain Networks
From www.nih.gov – Sep 3, 2013
Previous studies have shown that many people with schizophrenia have de novo, or new, genetic mutations. These misspellings in a gene’s DNA sequence
- occur spontaneously and so aren’t shared by their close relatives.
Dr. Mary-Claire King of the University of Washington in Seattle and colleagues set out to
- identify spontaneous genetic mutations in people with schizophrenia and
- to assess where and when in the brain these misspelled genes are turned on, or expressed.
The study was funded in part by NIH’s National Institute of Mental Health (NIMH). The results were published in the August 1, 2013, issue of Cell.
The researchers sequenced the exomes (protein-coding DNA regions) of 399 people—105 with schizophrenia plus their unaffected parents and siblings. Gene variations
that were found in a person with schizophrenia but not in either parent were considered spontaneous.
The likelihood of having a spontaneous mutation was associated with
- the age of the father in both affected and unaffected siblings.
Significantly more mutations were found in people
- whose fathers were 33-45 years at the time of conception compared to 19-28 years.
Among people with schizophrenia, the scientists identified
- 54 genes with spontaneous mutations
- predicted to cause damage to the function of the protein they encode.
The researchers used newly available database resources that show
- where in the brain and when during development genes are expressed.
The genes form an interconnected expression network with many more connections than
- that of the genes with spontaneous damaging mutations in unaffected siblings.
The spontaneously mutated genes in people with schizophrenia
- were expressed in the prefrontal cortex, a region in the front of the brain.
The genes are known to be involved in important pathways in brain development. Fifty of these genes were active
- mainly during the period of fetal development.
“Processes critical for the brain’s development can be revealed by the mutations that disrupt them,” King says. “Mutations can lead to loss of integrity of a whole pathway,
not just of a single gene.”
These findings support the concept that schizophrenia may result, in part, from
- disruptions in development in the prefrontal cortex during fetal development.
James E. Darnell’s “Reflections”
A brief history of the discovery of RNA and its role in transcription — peppered with career advice
By Joseph P. Tiano
James Darnell begins his Journal of Biological Chemistry “Reflections” article by saying, “graduate students these days
- have to swim in a sea virtually turgid with the daily avalanche of new information and
- may be momentarily too overwhelmed to listen to the aging.
I firmly believe how we learned what we know can provide useful guidance for how and what a newcomer will learn.” Considering his remarkable discoveries in
- RNA processing and eukaryotic transcriptional regulation
spanning 60 years of research, Darnell’s advice should be cherished. In his second year at medical school at Washington University School of Medicine in St. Louis, while
studying streptococcal disease in Robert J. Glaser’s laboratory, Darnell realized he “loved doing the experiments” and had his first “career advancement event.”
He and technician Barbara Pesch discovered that in vivo penicillin treatment killed streptococci only in the exponential growth phase and not in the stationary phase. These
results were published in the Journal of Clinical Investigation and earned Darnell an interview with Harry Eagle at the National Institutes of Health.
Darnell arrived at the NIH in 1956, shortly after Eagle shifted his research interest to developing his minimal essential cell culture medium, still used. Eagle, then studying cell metabolism, suggested that Darnell take up a side project on poliovirus replication in mammalian cells in collaboration with Robert I. DeMars. DeMars’ Ph.D.
adviser was also James Watson’s mentor, so Darnell met Watson, who invited him to give a talk at Harvard University, which led to an assistant professor position
at the MIT under Salvador Luria. A take-home message is to embrace side projects, because you never know where they may lead: this project helped to shape
his career.
Darnell arrived in Boston in 1961. Following the discovery of DNA’s structure in 1953, the world of molecular biology was turning to RNA in an effort to understand how
proteins are made. Darnell’s background in virology (it was discovered in 1960 that viruses used RNA to replicate) was ideal for the aim of his first independent lab:
exploring mRNA in animal cells grown in culture. While at MIT, he developed a new technique for purifying RNA along with making other observations
- suggesting that nonribosomal cytoplasmic RNA may be involved in protein synthesis.
When Darnell moved to Albert Einstein College of Medicine for full professorship in 1964, it was hypothesized that heterogenous nuclear RNA was a precursor to mRNA.
At Einstein, Darnell discovered RNA processing of pre-tRNAs and demonstrated for the first time
- that a specific nuclear RNA could represent a possible specific mRNA precursor.
In 1967 Darnell took a position at Columbia University, and it was there that he discovered (simultaneously with two other labs) that
- mRNA contained a polyadenosine tail.
The three groups all published their results together in the Proceedings of the National Academy of Sciences in 1971. Shortly afterward, Darnell made his final career move
four short miles down the street to Rockefeller University in 1974.
Over the next 35-plus years at Rockefeller, Darnell never strayed from his original research question: How do mammalian cells make and control the making of different
mRNAs? His work was instrumental in the collaborative discovery of
- splicing in the late 1970s and
- in identifying and cloning many transcriptional activators.
Perhaps his greatest contribution during this time, with the help of Ernest Knight, was
- the discovery and cloning of the signal transducers and activators of transcription (STAT) proteins.
And with George Stark, Andy Wilks and John Krowlewski, he described
- cytokine signaling via the JAK-STAT pathway.
Darnell closes his “Reflections” with perhaps his best advice: Do not get too wrapped up in your own work, because “we are all needed and we are all in this together.”

http://www.asbmb.org/assets/0/366/418/428/85528/85529/85530/8758cb87-84ff-42d6-8aea-96fda4031a1b.jpg
{kind=link}
Recent findings on presenilins and signal peptide peptidase
By Dinu-Valantin Bălănescu

Fig. 1 from the minireview shows a schematic depiction of γ-secretase and SPP
http://www.asbmb.org/assets/0/366/418/428/85528/85529/85530/c2de032a-daad-41e5-ba19-87a17bd26362.png
{kind=link}
GxGD proteases are a family of intramembranous enzymes capable of hydrolyzing
- the transmembrane domain of some integral membrane proteins.
The GxGD family is one of the three families of
- intramembrane-cleaving proteases discovered so far (along with the rhomboid and site-2 protease) and
- includes the γ-secretase and the signal peptide peptidase.
Although only recently discovered, a number of functions in human pathology and in numerous other biological processes
- have been attributed to γ-secretase and SPP.
Taisuke Tomita and Takeshi Iwatsubo of the University of Tokyo highlighted the latest findings on the structure and function of γ-secretase and SPP
in a recent minireview in The Journal of Biological Chemistry.
- γ-secretase is involved in cleaving the amyloid-β precursor protein, thus producing amyloid-β peptide,
the main component of senile plaques in Alzheimer’s disease patients’ brains. The complete structure of mammalian γ-secretase is not yet known; however,
Tomita and Iwatsubo note that biochemical analyses have revealed it to be a multisubunit protein complex.
- Its catalytic subunit is presenilin, an aspartyl protease.
In vitro and in vivo functional and chemical biology analyses have revealed that
- presenilin is a modulator and mandatory component of the γ-secretase–mediated cleavage of APP.
Genetic studies have identified three other components required for γ-secretase activity:
- nicastrin,
- anterior pharynx defective 1 and
- presenilin enhancer 2.
By coexpression of presenilin with the other three components, the authors managed to
- reconstitute γ-secretase activity.
Tomita and Iwatsubo determined using the substituted cysteine accessibility method and by topological analyses, that
- the catalytic aspartates are located at the center of the nine transmembrane domains of presenilin,
- by revealing the exact location of the enzyme’s catalytic site.
The minireview also describes in detail the formerly enigmatic mechanism of γ-secretase mediated cleavage.
SPP, an enzyme that cleaves remnant signal peptides in the membrane
- during the biogenesis of membrane proteins and
- signal peptides from major histocompatibility complex type I,
- also is involved in the maturation of proteins of the hepatitis C virus and GB virus B.
Bioinformatics methods have revealed in fruit flies and mammals four SPP-like proteins,
- two of which are involved in immunological processes.
By using γ-secretase inhibitors and modulators, it has been confirmed
- that SPP shares a similar GxGD active site and proteolytic activity with γ-secretase.
Upon purification of the human SPP protein with the baculovirus/Sf9 cell system,
- single-particle analysis revealed further structural and functional details.
HLA targeting efficiency correlates with human T-cell response magnitude and with mortality from influenza A infection
From www.pnas.org – Sep 3, 2013 4:24 PM
Experimental and computational evidence suggests that
- HLAs preferentially bind conserved regions of viral proteins, a concept we term “targeting efficiency,” and that
- this preference may provide improved clearance of infection in several viral systems.
To test this hypothesis, T-cell responses to A/H1N1 (2009) were measured from peripheral blood mononuclear cells obtained from a household cohort study
performed during the 2009–2010 influenza season. We found that HLA targeting efficiency scores significantly correlated with
- IFN-γ enzyme-linked immunosorbent spot responses (P = 0.042, multiple regression).
A further population-based analysis found that the carriage frequencies of the alleles with the lowest targeting efficiencies, A*24,
- were associated with pH1N1 mortality (r = 0.37, P = 0.031) and
- are common in certain indigenous populations in which increased pH1N1 morbidity has been reported.
HLA efficiency scores and HLA use are associated with CD8 T-cell magnitude in humans after influenza infection.
The computational tools used in this study may be useful predictors of potential morbidity and
- identify immunologic differences of new variant influenza strains
- more accurately than evolutionary sequence comparisons.
Population-based studies of the relative frequency of these alleles in severe vs. mild influenza cases
- might advance clinical practices for severe H1N1 infections among genetically susceptible populations.
Metabolomics in drug target discovery
J D Rabinowitz et al.
Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, NJ.
Cold Spring Harbor Symposia on Quantitative Biology 11/2011; 76:235-46.
http://dx.doi.org:/10.1101/sqb.2011.76.010694
Most diseases result in metabolic changes. In many cases, these changes play a causative role in disease progression. By identifying pathological metabolic changes,
- metabolomics can point to potential new sites for therapeutic intervention.
Particularly promising enzymatic targets are those that
- carry increased flux in the disease state.
Definitive assessment of flux requires the use of isotope tracers. Here we present techniques for
- finding new drug targets using metabolomics and isotope tracers.
The utility of these methods is exemplified in the study of three different viral pathogens. For influenza A and herpes simplex virus,
- metabolomic analysis of infected versus mock-infected cells revealed
- dramatic concentration changes around the current antiviral target enzymes.
Similar analysis of human-cytomegalovirus-infected cells, however, found the greatest changes
- in a region of metabolism unrelated to the current antiviral target.
Instead, it pointed to the tricarboxylic acid (TCA) cycle and
- its efflux to feed fatty acid biosynthesis as a potential preferred target.
Isotope tracer studies revealed that cytomegalovirus greatly increases flux through
- the key fatty acid metabolic enzyme acetyl-coenzyme A carboxylase.
- Inhibition of this enzyme blocks human cytomegalovirus replication.
Examples where metabolomics has contributed to identification of anticancer drug targets are also discussed. Eventual proof of the value of
- metabolomics as a drug target discovery strategy will be
- successful clinical development of therapeutics hitting these new targets.
Related References
Use of metabolic pathway flux information in targeted cancer drug design. Drug Discovery Today: Therapeutic Strategies 1:435-443, 2004.
Detection of resistance to imatinib by metabolic profiling: clinical and drug development implications. Am J Pharmacogenomics. 2005;5(5):293-302. Review. PMID: 16196499
Medicinal chemistry, metabolic profiling and drug target discovery: a role for metabolic profiling in reverse pharmacology and chemical genetics.
Mini Rev Med Chem. 2005 Jan;5(1):13-20. Review. PMID: 15638788 [PubMed – indexed for MEDLINE] Related citations
Development of Tracer-Based Metabolomics and its Implications for the Pharmaceutical Industry. Int J Pharm Med 2007; 21 (3): 217-224.
Use of metabolic pathway flux information in anticancer drug design. Ernst Schering Found Symp Proc. 2007;(4):189-203. Review. PMID: 18811058
Pharmacological targeting of glucagon and glucagon-like peptide 1 receptors has different effects on energy state and glucose homeostasis in diet-induced obese mice. J Pharmacol Exp Ther. 2011 Jul;338(1):70-81.http://dx.doi.org:/10.1124/jpet.111.179986. PMID: 21471191
Single valproic acid treatment inhibits glycogen and RNA ribose turnover while disrupting glucose-derived cholesterol synthesis in liver as revealed by the
[U-C(6)]-d-glucose tracer in mice. Metabolomics. 2009 Sep;5(3):336-345. PMID: 19718458
Metabolic Pathways as Targets for Drug Screening, Metabolomics, Dr Ute Roessner (Ed.), ISBN: 978-953-51-0046-1, InTech, Available from: http://www.intechopen.com/books/metabolomics/metabolic-pathways-as-targets-for-drug-screening
Iron regulates glucose homeostasis in liver and muscle via AMP-activated protein kinase in mice. FASEB J. 2013 Jul;27(7):2845-54.
http://dx.doi.org:/10.1096/fj.12-216929. PMID: 23515442
Metabolomics and systems pharmacology: why and how to model the human metabolic network for drug discovery
Drug Discov. Today 19 (2014), 171–182 http://dx.doi.org:/10.1016/j.drudis.2013.07.014
Highlights
- We now have metabolic network models; the metabolome is represented by their nodes.
- Metabolite levels are sensitive to changes in enzyme activities.
- Drugs hitchhike on metabolite transporters to get into and out of cells.
- The consensus network Recon2 represents the present state of the art, and has predictive power.
- Constraint-based modelling relates network structure to metabolic fluxes.
Metabolism represents the ‘sharp end’ of systems biology, because changes in metabolite concentrations are
- necessarily amplified relative to changes in the transcriptome, proteome and enzyme activities, which can be modulated by drugs.
To understand such behaviour, we therefore need (and increasingly have) reliable consensus (community) models of
- the human metabolic network that include the important transporters.
Small molecule ‘drug’ transporters are in fact metabolite transporters, because
- drugs bear structural similarities to metabolites known from the network reconstructions and
- from measurements of the metabolome.
Recon2 represents the present state-of-the-art human metabolic network reconstruction; it can predict inter alia:
(i) the effects of inborn errors of metabolism;
(ii) which metabolites are exometabolites, and
(iii) how metabolism varies between tissues and cellular compartments.
However, even these qualitative network models are not yet complete. As our understanding improves
- so do we recognise more clearly the need for a systems (poly)pharmacology.
Introduction – a systems biology approach to drug discovery
It is clearly not news that the productivity of the pharmaceutical industry has declined significantly during recent years
- following an ‘inverse Moore’s Law’, Eroom’s Law, or
- that many commentators, consider that the main cause of this is
- because of an excessive focus on individual molecular target discovery rather than a more sensible strategy
- based on a systems-level approach (Fig. 1).

Figure 1.
The change in drug discovery strategy from ‘classical’ function-first approaches (in which the assay of drug function was at the tissue or organism level),
with mechanistic studies potentially coming later, to more-recent target-based approaches where initial assays usually involve assessing the interactions
of drugs with specified (and often cloned, recombinant) proteins in vitro. In the latter cases, effects in vivo are assessed later, with concomitantly high levels of attrition.
Arguably the two chief hallmarks of the systems biology approach are:
(i) that we seek to make mathematical models of our systems iteratively or in parallel with well-designed ‘wet’ experiments, and
(ii) that we do not necessarily start with a hypothesis but measure as many things as possible (the ’omes) and
- let the data tell us the hypothesis that best fits and describes them.
Although metabolism was once seen as something of a Cinderella subject,
- there are fundamental reasons to do with the organisation of biochemical networks as
- to why the metabol(om)ic level – now in fact seen as the ‘apogee’ of the ’omics trilogy –
- is indeed likely to be far more discriminating than are
- changes in the transcriptome or proteome.
The next two subsections deal with these points and Fig. 2 summarises the paper in the form of a Mind Map.

http://ars.els-cdn.com/content/image/1-s2.0-S1359644613002481-gr2.jpg
{kind=link}
Metabolic Disease Drug Discovery— “Hitting the Target” Is Easier Said Than Done
David E. Moller, et al. http://dx.doi.org:/10.1016/j.cmet.2011.10.012
Despite the advent of new drug classes, the global epidemic of cardiometabolic disease has not abated. Continuing
- unmet medical needs remain a major driver for new research.
Drug discovery approaches in this field have mirrored industry trends, leading to a recent
- increase in the number of molecules entering development.
However, worrisome trends and newer hurdles are also apparent. The history of two newer drug classes—
- glucagon-like peptide-1 receptor agonists and
- dipeptidyl peptidase-4 inhibitors—
illustrates both progress and challenges. Future success requires that researchers learn from these experiences and
- continue to explore and apply new technology platforms and research paradigms.
The global epidemic of obesity and diabetes continues to progress relentlessly. The International Diabetes Federation predicts an even greater diabetes burden (>430 million people afflicted) by 2030, which will disproportionately affect developing nations (International Diabetes Federation, 2011). Yet
- existing drug classes for diabetes, obesity, and comorbid cardiovascular (CV) conditions have substantial limitations.
Currently available prescription drugs for treatment of hyperglycemia in patients with type 2 diabetes (Table 1) have notable shortcomings. In general,
- their efficacy profiles fall short of achieving accepted treatment goals (American Diabetes Association, 2011).
Therefore, clinicians must often use combination therapy, adding additional agents over time. Ultimately many patients will need to use insulin—a therapeutic class first introduced in 1922. Most existing agents also have
- issues around safety and tolerability as well as dosing convenience (which can impact patient compliance).
Pharmacometabolomics, also known as pharmacometabonomics, is a field which stems from metabolomics,
- the quantification and analysis of metabolites produced by the body.
It refers to the direct measurement of metabolites in an individual’s bodily fluids, in order to
- predict or evaluate the metabolism of pharmaceutical compounds, and
- to better understand the pharmacokinetic profile of a drug.
Alternatively, pharmacometabolomics can be applied to measure metabolite levels
- following the administration of a pharmaceutical compound, in order to
- monitor the effects of the compound on certain metabolic pathways(pharmacodynamics).
This provides detailed mapping of drug effects on metabolism and
- the pathways that are implicated in mechanism of variation of response to treatment.
In addition, the metabolic profile of an individual at baseline (metabotype) provides information about
- how individuals respond to treatment and highlights heterogeneity within a disease state.
All three approaches require the quantification of metabolites found
- in bodily fluids and tissue, such as blood or urine, and
- can be used in the assessment of pharmaceutical treatment options for numerous disease states.

http://upload.wikimedia.org/wikipedia/commons/thumb/e/eb/OMICS.png/350px-OMICS.png
{kind=link}
Pharmacometabolomics is thought to provide information that
- complements that gained from other omics, namely genomics, transcriptomics, and proteomics.
Looking at the characteristics of an individual down through these different levels of detail, there is an
- increasingly more accurate prediction of a person’s ability to respond to a pharmaceutical compound.
- the genome, made up of 25 000 genes, can indicate possible errors in drug metabolism;
- the transcriptome, made up of 85,000 transcripts, can provide information about which genes important in metabolism are being actively transcribed;
- and the proteome, >10,000,000 members, depicts which proteins are active in the body to carry out these functions.
Pharmacometabolomics complements the omics with
- direct measurement of the products of all of these reactions, but with perhaps a relatively
- smaller number of members: that was initially projected to be approximately 2200 metabolites,
but could be a larger number when gut derived metabolites and xenobiotics are added to the list. Overall, the goal of pharmacometabolomics is
- to more closely predict or assess the response of an individual to a pharmaceutical compound,
- permitting continued treatment with the right drug or dosage
- depending on the variations in their metabolism and ability to respond to treatment.
Pharmacometabolomic analyses, through the use of a metabolomics approach,
- can provide a comprehensive and detailed metabolic profile or “metabolic fingerprint” for an individual patient.
Such metabolic profiles can provide a complete overview of individual metabolite or pathway alterations,
- providing a more realistic depiction of disease phenotypes.
This approach can then be applied to the prediction of response to a pharmaceutical compound
- by patients with a particular metabolic profile.
Pharmacometabolomic analyses of drug response are
- often coupled or followed up with pharmacogenetics studies.
Pharmacogenetics focuses on the identification of genetic variations (e.g. single-nucleotide polymorphisms)
- within patients that may contribute to altered drug responses and overall outcome of a certain treatment.
The results of pharmacometabolomics analyses can act to “inform” or “direct”
- pharmacogenetic analyses by correlating aberrant metabolite concentrations or metabolic pathways to potential alterations at the genetic level.
This concept has been established with two seminal publications from studies of antidepressants serotonin reuptake inhibitors
- where metabolic signatures were able to define a pathway implicated in response to the antidepressant and
- that lead to identification of genetic variants within a key gene
- within the highlighted pathway as being implicated in variation in response.
These genetic variants were not identified through genetic analysis alone and hence
- illustrated how metabolomics can guide and inform genetic data.
en.wikipedia.org/wiki/Pharmacometabolomics
Benznidazole Biotransformation and Multiple Targets in Trypanosoma cruzi Revealed by Metabolomics
Andrea Trochine, Darren J. Creek, Paula Faral-Tello, Michael P. Barrett, Carlos Robello
Published: May 22, 2014 http://dx.doi.org:/10.1371/journal.pntd.0002844
The first line treatment for Chagas disease, a neglected tropical disease caused by the protozoan parasite Trypanosoma cruzi,
- involves administration of benznidazole (Bzn).
Bzn is a 2-nitroimidazole pro-drug which requires nitroreduction to become active. We used a
- non-targeted MS-based metabolomics approach to study the metabolic response of T. cruzi to Bzn.
Parasites treated with Bzn were minimally altered compared to untreated trypanosomes, although the redox active thiols
- trypanothione,
- homotrypanothione and
- cysteine
were significantly diminished in abundance post-treatment. In addition, multiple Bzn-derived metabolites were detected after treatment.
These metabolites included reduction products, fragments and covalent adducts of reduced Bzn
- linked to each of the major low molecular weight thiols:
- trypanothione,
- glutathione,
- g-glutamylcysteine,
- glutathionylspermidine,
- cysteine and
- ovothiol A.
Bzn products known to be generated in vitro by the unusual trypanosomal nitroreductase, TcNTRI,
- were found within the parasites,
- but low molecular weight adducts of glyoxal, a proposed toxic end-product of NTRI Bzn metabolism, were not detected.
Our data is indicative of a major role of the
- thiol binding capacity of Bzn reduction products
- in the mechanism of Bzn toxicity against T. cruzi.
Prefacing the e-Book Epilogue: Metabolic Genomics and Pharmaceutics
Author and Curator: Larry H. Bernstein, MD, FCAP
Adieu, adieu, adieu …
Sound of Music

This work has been a coming to terms with my scientific and medical end of career balancing in a difficult time after retiring, but it has been rewarding. In the clinical laboratories, radiology, anesthesiology, and in pharmacy, there has been some significant progress in support of surgical, gynecological, developmental, medical practices, and even neuroscience directed disciplines, as well as epidemiology over a period of half a century. Even then, cancer and neurological diseases have been most difficult because the scientific basic research has either not yet uncovered a framework, or because that framework has proved to be multidimensional. In the clinical laboratory sciences, there has been enormous progress in instrumental analysis, with the recent opening of molecular methods not yet prepared for routine clinical use, which will be a very great challenge to the profession, which has seen the development of large sample volume, multianalite, high-throughput, low-cost support emerging for decades. The capabilities now underway will also enrrich the the capabilities of the anatomic pathology suite and the capabilities of 3-dimensional radiological examination. In both pathology and radiology, we have seen the division of the fields into major subspecialties. The development of the electronic health record had to take lessons from the first developments in the separate developments of laboratory, radiology, and pharmacy health record systems, to which were added, full cardiology monitoring systems. These have been unintegrated. This made it difficult to bring forth a suitable patient health record because the information needed to support decision-making by practitioners was in separate “silos”. The mathematical methods that are being applied to the -OMICS sciences, can be brought to bear on the simplification and amplification of the clinicians’ ability to make decisions with near “errorless” discrimination, still allowing for an element of “art” in carrying out the history, physical examination, and knowledge unique to every patient.
We are at this time opening a very large, complex, study of biology in relationship to the human condition. This will require sufficient resources to be invested in the development of these for a better society, which I suspect, will go on beyond the life of my grandchildren. Hopefully, the long-term dangers of climate change will be controlled in that time. As a society, or as a group of interdependent societies, we have no long term interest in continuing self-destructive behaviors that have predominated in the history of mankind. I now top off these discussions with some further elucidation of what lies before us.
Metabolomics and systems pharmacology: why and how to model the human metabolic network for drug discovery
Douglas B. Kell and Royston Goodacre
School of Chemistry and Manchester Institute of Biotechnology, The University of Manchester, Manchester, UK
Drug Discovery Today Feb 2014;19(2) http://dx.doi.org/10.1016/j.drudis.2013.07.014
Metabolism represents the ‘sharp end’ of systems biology,
- because changes in metabolite concentrations
- are necessarily amplified relative to
- changes in the transcriptome, proteome and enzyme activities,
- which can be modulated by drugs.
To understand such behaviour, we therefore need
(and increasingly have)
- reliable consensus (community) models of the human metabolic network
- that include the important transporters.
Small molecule ‘drug’ transporters are in fact metabolite transporters,
- because drugs bear structural similarities to metabolites known
- from the network reconstructions and from measurements of the metabolome.
Recon2 represents the present state-of-the-art human metabolic
network reconstruction; it can predict inter alia:
- the effects of inborn errors of metabolism;
- which metabolites are exometabolites, and
- how metabolism varies between tissues and cellular compartments.
Even these qualitative network models are not yet complete. As our
understanding improves so do we recognize more clearly the need for a systems (poly)pharmacology.
Modelling biochemical networks – why we do so
There are at least four types of reasons as to why one would wish to model a biochemical network:
- Assessing whether the model is accurate, in the sense that it
reflects – or can be made to reflect – known experimental facts. - Establishing what changes in the model would improve the
consistency of its behaviour with experimental observations
and improved predictability, such as with respect to metabolite
concentrations or fluxes. - Analyzing the model, typically by some form of sensitivity
analysis, to understand which parts of the system contribute
most to some desired functional properties of interest. - Hypothesis generation and testing, enabling one to analyse
rapidly the effects of manipulating experimental conditions in
the model without having to perform complex and costly
experiments (or to restrict the number that are performed).
In particular, it is normally considerably cheaper to perform
studies of metabolic networks in silico before trying a smaller
number of possibilities experimentally; indeed for combinatorial
reasons it is often the only approach possible. Although
our focus here is on drug discovery, similar principles apply to the
modification of biochemical networks for purposes of ‘industrial’
or ‘white’ biotechnology.
Why we choose to model metabolic networks more than
- transcriptomic or proteomic networks
comes from the recognition – made particularly clear
- by workers in the field of metabolic control analysis
– that, although changes in the activities of individual enzymes tend to have
- rather small effects on metabolic fluxes,
- they can and do have very large effects on metabolite concentrations (i.e. the metabolome).
Modelling biochemical networks – how we do so
Although one could seek to understand the
- time-dependent spatial distribution of signalling and metabolic substances within indivi
dual cellular compartments and - while spatially discriminating analytical methods such as Raman spectroscopy and
mass spectrometry do exist for the analysis of drugs in situ,
- the commonest type of modelling, as in the spread of substances in
ecosystems, - assumes ‘fully mixed’ compartments and thus ‘pools’ of metabolites.
Although an approximation, this ‘bulk’ modelling will be necessary for complex ecosystems such as humans where, in addition to the need for tissue- and cell-specific models, microbial communities inhabit this superorganism and the
gut serves as a source for nutrients courtesy of these symbionts.
Topology and stoichiometry of metabolic networks as major constraints on fluxes
Given their topology, which admits a wide range of parameters for
delivering the same output effects and thereby reflects biological
robustness,
- metabolic networks have two especially important constraints that assist their accurate modelling:
(i) the conservation of mass and charge, and
(ii) stoichiometric and thermodynamic constraints.
These are tighter constraints than apply to signalling networks.
New developments in modelling the human metabolic network
Since 2007, several groups have been developing improved but nonidentical models of the human metabolic network at a generalised level and in tissue-specific forms. Following a similar community-driven strategy in Saccharomyces cerevisiae, surprisingly similar to humans, and in Salmonella typhimurium,
we focus in particular on a recent consensus paper that provides a highly curated and semantically annotated model of the human metabolic network, termed
- Recon2 (http://humanmetabolism.org/).
In this work, a substantial number of the major groups active in this area came together to provide a carefully and manually constructed/curated network, consisting of some 1789 enzyme-encoding genes, 7440 reactions and 2626 unique metabolites distributed over eight cellular compartments. A variety of dead-end metabolites and blocked reactions remain (essentially orphans and widows). But Recon2 was able to
- account for some 235 inborn errors of metabolism,
- a variety of metabolic ‘tasks’ (defined as a non-zero flux through a reaction or through a pathway leading to the production of a metabolite Q from a metabolite P).
- filtering based on expression profiling allowed the construction of 65 cell-type-specific models.
- Excreted or exometabolites are an interesting set of metabolites,
- and Recon2 could predict successfully a substantial fraction of those
Role of transporters in metabolic fluxes
The uptake and excretion of metabolites between cells and their macrocompartments
- requires specific transporters and in the order of one third of ‘metabolic’ enzymes,
- and indeed of membrane proteins, are in fact transporters or equivalent.
What is of particular interest (to drug discovery), based on their structural similarities, is the increasing recognition (Fig. 3) that pharmaceutical drugs also
- get into and out of cells by ‘hitchhiking’ on such transporters, and not –
to any significant extent –
- by passing through phospholipid bilayer portions
of cellular membranes.
This makes drug discovery even more a problem of systems biology than of biophysics.

Two views of the role of solute carriers and other transporters in cellular drug uptake. (a) A more traditional view in which all so-called ‘passive’drug uptake occurs through any unperturbed bilayer portion of membrane that might be present.
(b) A view in which the overwhelming fraction of drug is taken up via solute transporters or other carriers that are normally used for the uptake of intermediary metabolites. Noting that the protein:lipid ratio of biomembranes is typically 3:1 to 1:1 and that proteins vary in mass and density (a typical density is 1.37 g/ml) as does their extension, for example, normal to the ca. 4.5 nm lipid bilayer region, the figure attempts to portray a section of a membrane with realistic or typical sizes and amounts of proteins and lipids. Typical protein areas when viewed normal to the membrane are 30%, membranes are rather more ‘mosaic’ than ‘fluid’ and there is some evidence that there might be no genuinely ‘free’ bulk lipids (typical phospholipid masses are 750 Da) in biomembranes that are uninfluenced by proteins. Also shown is a typical drug: atorvastatin (LipitorW) – with a molecular mass of 558.64 Da – for size comparison purposes. If proteins are modelled as
cylinders, a cylinder with a diameter of 3.6 nm and a length of 6 nm has a molecular mass of ca. 50 kDa. Note of course that in a ‘static’ picture we cannot show the dynamics of either phospholipid chains or lipid or protein diffusion.
‘Newly discovered’ metabolites and/or their roles
To illustrate the ‘unfinished’ nature even of Recon2, which concentrates on the metabolites created via enzymes encoded in the human genome, and leaving aside the more exotic metabolites of drugs and foodstuffs and the ‘secondary’ metabolites of microorganisms, there are several examples of interesting ‘new’ (i.e. more or less recently recognised) human metabolites or roles thereof that are worth highlighting, often from studies seeking biomarkers of various diseases – for caveats of biomarker discovery, which is not a topic that we are covering here, and the need for appropriate experimental design. In addition, classes of metabolites not well represented in Recon2 are oxidised molecules such as those caused by nonenzymatic reaction of metabolites with free radicals such as the hydroxyl radical generated by unliganded iron. There is also significant interest in using methods of determining small molecules such as those in the
metabolome (inter alia) for assessing the ‘exposome’, in other words all the potentially polluting agents to which an
individual has been exposed.
Recently discovered effects of metabolites on enzymes
Another combinatorial problem reflects the fact that in molecular enzymology it is not normally realistic to assess every possible metabolite to determine whether it is an effector (i.e.activator or inhibitor) of the enzyme under study. Typical proteins are highly promiscuous and there is increasing evidence for the comparative promiscuity of metabolites
and pharmaceutical drugs. Certainly the contribution of individual small effects of multiple parameter changes can have substantial effects on the potential flux through an overall pathway, which makes ‘bottom up’ modelling an inexact science. Even merely mimicking the vivo (in Escherichia coli) concentrations of K+, Na+, Mg2+, phosphate, glutamate, sulphate and Cl significantly modulated the activities of several enzymes tested relative to the ‘usual’ assay conditions. Consequently, we need to be alive to the possibility of many (potentially major) interactions of which we are as yet ignorant. One class of example relates to the effects of the very widespread post-translational modification on metabolic
enzyme activities.
A recent and important discovery (Fig. 4) is that a single transcriptome experiment, serving as a surrogate for fluxes through individual steps, provides a huge constraint on possible models, and predicts in a numerically tractable way and
with much improved accuracy the fluxes to exometabolites without the need for such a variable ‘biomass’ term. Other recent and related strategies that exploit modern advances in ‘omics and network biology to limit the search space in constraint-based metabolic modelling.
Fig 4. Workflow for expression-profile-constrained metabolic flux estimation
- Genome-scale metabolic model with gene-protein-reaction relationships
- Map absolute gene expression levels to reactions
- Maximise correlation between absolute gene expression and metabolic flux
- Predict fluxes to exometabolites
- Compare predicted with experimental fluxes to exometabolites
Drug Discovery Today
The steps in a workflow that uses constraints based on (i) metabolic network stoichiometry and chemical reaction properties (both encoded in the model) plus, and (ii) absolute (RNA-Seq) transcript expression profiles to enable the
accurate modelling of pathway and exometabolite fluxes. .
Concluding remarks – the role of metabolomics in systems pharmacology
What is becoming increasingly clear, as we recognize that to understand living organisms in health and disease we must treat them as systems, is that we must bring together our knowledge of the topologies and kinetics of metabolic networks with our knowledge of the metabolite concentrations (i.e. metabolomes) and fluxes. Because of the huge constraints imposed on metabolism by reaction stoichiometries, mass conservation and thermodynamics, comparatively few well-chosen ‘omics measurements might be needed to do this reliably (Fig. 4). Indeed, a similar approach exploiting constraints has come to the fore in denovo protein folding and interaction studies.
What this leads us to in drug discovery is the need to develop and exploit a ‘systems pharmacology’ where multiple binding targets are chosen purposely and simultaneously. Along with other measures such as phenotypic screening, and the integrating of the full suite of e-science approaches, one can anticipate considerable improvements in the rate of discovery of safe and effective drugs.
Metabolomics: the apogee of the omics trilogy
Gary J.!Patti, Oscar Yanes and Gary Siuzdak
Metabolites, the chemical entities that are transformed during metabolism, provide a functional readout of cellular biochemistry. With emerging technologies in mass spectrometry, thousands of metabolites can now be
quantitatively measured from minimal amounts of biological material, which has thereby enabled systems-level analyses. By performing global metabolite profiling, also known as untargeted metabolomics, new discoveries linking cellular pathways to biological mechanism are being revealed and are shaping our understanding of cell biology, physiology and medicine.
Metabolites are small molecules that are chemically transformed during metabolism and, as such, they provide a functional readout of cellular state. Unlike genes and proteins, the functions of which are subject to epigenetic regulation and posttranslational modifications, respectively, metabolites serve as direct signatures of biochemical activity and are therefore easier to correlate with phenotype. In this context, metabolite profiling, or metabolomics, has become a powerful approach that has been widely adopted for clinical diagnostics.
The field of metabolomics has made remarkable progress within the past decade and has implemented new tools that have offered mechanistic insights by allowing for the correlation of biochemical changes with phenotype.
In this Innovation article, we first define and differentiate between the targeted and untargeted approaches to metabolomics. We then highlight the value of untargeted metabolomics in particular and outline a guide to performing such studies. Finally, we describe selected applications of un targeted metabolomics and discuss their potential in cell biology.
- metabolites serve as direct signatures of biochemical activity
- In some instances, it may be of interest to examine a defined set of metabolites by using a targeted approach.
- In other cases, an untargeted or global approach may be taken in which as many metabolites as possible are measured and compared between samples without bias.
- Ultimately, the number and chemical composition of metabolites to be studied is a defining attribute of any metabolomic experiment and shapes experimental design with respect to sample preparation and choice of instrumentation.
The targeted and untargeted workflow for LC/MS-based metabolomics.
a | In the triple quadrupole (QqQ)-based targeted metabolomic workflow, standard compounds for the metabolites of interest are first used to set up selected reaction monitoring methods. Here, optimal instrument voltages are determined and response curves are generated for absolute quantification. After the targeted methods have been established
on the basis of standard metabolites, metabolites are extracted from tissues, biofluids or cell cultures and analysed. The data output provides quantification only of those metabolites for which standard methods have been built.
b | In the untargeted metabolomic workflow, metabolites are first isolated from biological samples and subsequently analysed by liquid chromatography followed by mass spectrometry (LC/MS). After data acquisition, the results are processed by using bioinformatic software such as XCMS to perform nonlinear retention time alignment and identify peaks that are changing between the groups of samples measured. The m/z value s for the peaks of interest are searched in metabolite databases to obtain putative identifications. Putative identifications are then confirmed
by comparing tandem mass spectrometry (MS/MS) data and retention time data to that of standard compounds. The untargeted workflow is global in scope and outputs data related to comprehensive cellular metabolism.
Metabolic Biomarker and Kinase Drug Target Discovery in Cancer Using Stable Isotope-Based Dynamic Metabolic Profiling (SIDMAP)
László G. Boros1*, Daniel J. Brackett2 and George G. Harrigan3
1UCLA School of Medicine, Harbor-UCLA Research and Education Institute, Torrance, CA. 2Department of Surgery, University of Oklahoma Health Sciences Center & VA Medical Center, Oklahoma City, OK, 3Global High Throughput
Screening (HTS), Pharmacia Corporation, Chesterfield, MO.
Current Cancer Drug Targets, 2003, 3, 447-455.
Tumor cells respond to growth signals by the activation of protein kinases, altered gene expression and significant modifications in substrate flow and redistribution among biosynthetic pathways. This results in a proliferating phenotype
with altered cellular function. These transformed cells exhibit unique anabolic characteristics, which includes increased and preferential utilization of glucose through the non-oxidative steps of the pentose cycle for nucleic acid synthesis but limited denovo fatty acid synthesis and TCA cycle glucose oxidation. This primarily nonoxidative anabolic profile reflects an undifferentiated highly proliferative aneuploid cell phenotype and serves as a reliable metabolic biomarker to determine cell proliferation rate and the level of cell transformation/differentiation in response to drug treatment. Novel drugs effective in particular cancers exert their anti-proliferative effects by inducing significant reversions of a few specific non-oxidative anabolic pathways. Here we present evidence that cell transformation of various mechanisms is sustained by a unique
disproportional substrate distribution between the two branches of the pentose cycle for nucleic acid synthesis, glycolysis and the TCA cycle for fatty acid synthesis and glucose oxidation. This can be demonstrated by the broad labeling and unique specificity of [1,2-13C2]glucose to trace a large number of metabolites in the metabolome. Stable isotope-based dynamic metabolic profiles (SIDMAP) serve the drug discovery process by providing a powerful new tool that integrates the metabolome into a functional genomics approach to developing new drugs. It can be used in screening kinases and their metabolic targets, which can therefore be more efficiently characterized, speeding up and improving drug testing, approval and labeling processes by saving trial and error type study costs in drug testing.
Navigating the HumanMetabolome for Biomarker Identification and Design of Pharmaceutical Molecules
Irene Kouskoumvekaki and Gianni Panagiotou
Department of Systems Biology, Center for Biological Sequence Analysis, Building 208, Technical University of Denmark, Lyngby, Denmark
Hindawi Publishing Corporation Journal of Biomedicine and Biotechnology 2011, Article ID 525497, 19 pages
http://dx.doi.org:/10.1155/2011/525497
Metabolomics is a rapidly evolving discipline that involves the systematic study of endogenous small molecules that characterize the metabolic pathways of biological systems. The study of metabolism at a global level has the potential to contribute significantly to biomedical research, clinical medical practice, as well as drug discovery. In this paper, we present the most up-to-date metabolite and metabolic pathway resources, and we summarize the statistical, and machine-learning tools used for the analysis of data from clinical metabolomics.
Through specific applications on cancer, diabetes, neurological and other diseases, we demonstrate how these tools can facilitate diagnosis and identification of potential biomarkers for use within disease diagnosis. Additionally, we
discuss the increasing importance of the integration of metabolomics data in drug discovery. On a case-study based on the Human Metabolome Database (HMDB) and the Chinese Natural Product Database (CNPD), we demonstrate the close relatedness of the two data sets of compounds, and we further illustrate how structural similarity with human metabolites could assist in the design of novel pharmaceuticals and the elucidation of the molecular mechanisms of medicinal plants.
Metabolites are the byproducts of metabolism, which is itself the process of converting food energy to mechanical energy
or heat. Experts believe there are at least 3,000 metabolites that are essential for normal growth and development (primary metabolites) and thousands more unidentified (around 20,000, compared to an estimated 30,000 genes and 100,000 proteins) that are not essential for growth and development (secondary metabolites) but could represent prognostic, diagnostic, and surrogate markers for a disease state and a deeper understanding of mechanisms of disease.
Metabolomics, the study of metabolism at the global level, has the potential to contribute significantly to biomedical
research, and ultimately to clinical medical practice. It is a close counterpart to the genome, the transcriptome and the proteome. Metabolomics, genomics, proteomics, and other “-omics” grew out of the Human Genome Project, a massive research effort that began in the mid-1990s and culminated in 2003 with a complete mapping of all the genes in the human body. When discussing the clinical advantages of metabolomics, scientists point to the “real-world” assessment
of patient physiology that the metabolome provides since it can be regarded as the end-point of the “-omics” cascade. Other functional genomics technologies do not necessarily predict drug effects, toxicological response, or disease states at the phenotype but merely indicate the potential cause for phenotypical response. Metabolomics can bridge this information gap. The identification and measurement of metabolite profile dynamics of host changes provides the closest link to the various phenotypic responses. Thus it is clear that the global mapping of metabolic signatures pre- and postdrug treatment is a promising approach to identify possible functional relationships between medication and medical phenotype.
Human Metabolome Database (HMDB). Focusing on quantitative, analytic, or molecular scale information about metabolites, the enzymes and transporters associated with them, as well as disease related properties the HMDB represents the most complete bioinformatics and chemoinformatics medical information database. It contains records for thousands of endogenous metabolites identified by literature surveys (PubMed, OMIM, OMMBID, text books), data mining (KEGG, Metlin, BioCyc) or experimental analyses performed on urine, blood, and cerebrospinal fluid samples.
The annotation effort is aided by chemical parameter calculators and protein annotation tools originally developed for DrugBank.
A key feature that distinguishes the HMDB from other metabolic resources is its extensive support for higher level database searching and selecting functions. More than 175 hand-drawn-zoomable, fully hyperlinked human metabolic pathway maps can be found in HMDB and all these maps are quite specific to human metabolism and explicitly show the subcellular compartments where specific reactions are known to take place. As an equivalent to BLAST the HMDB contains a structure similarity search tool for chemical structures and users may sketch or paste a SMILES string of a query compound into the Chem-Query window. Submitting the query launches a structure similarity search tool that looks for common substructures from the query compound that match the HMDB’s metabolite database. The wealth of information and especially the extensive linkage to metabolic diseases
to normal and abnormal metabolite concentration ranges, to mutation/SNP data and to the genes, enzymes, reactions and pathways associated with many diseases of interest makes the HMDB one the most valuable tool in the hands of clinical chemists, nutritionists, physicians and medical geneticists.
Metabolomics in Drug Discovery and Polypharmacology Studies
Drug molecules generally act on specific targets at the cellular level, and upon binding to the receptors, they exert a desirable alteration of the cellular activities, regarded as the pharmaceutical effect. Current drug discovery depends largely on ransom screening, either high-throughput screening (HTS) in vitro, or virtual screening (VS) in silico. Because the number of available compounds is huge, several druglikeness filters are proposed to reduce the number of compounds that need to be evaluated. The ability to effectively predict if a chemical compound is “drug-like” or “nondruglike” is, thus, a valuable tool in the design, optimization, and selection of drug candidates for development. Druglikeness is a general descriptor of the potential of a small molecule to become a drug. It is not a unified descriptor
but a global property of a compound processing many specific characteristics such as good solubility, membrane permeability, half-life, and having a pharmacophore pattern to interact specifically with a target protein. These characteristics can be reflected as molecular descriptors such as molecular weight, log P, the number of hydrogen bond donors, the number of hydrogen-bond acceptors, the number of rotatable bonds, the number of rigid bonds, the number of rings in a molecule, and so forth.
Metabolomics for the Study of Polypharmacology of Natural Compounds
Internationally, there is a growing and sustained interest from both pharmaceutical companies and public in medicine from natural sources. For the public, natural medicine represent a holistic approach to disease treatment, with potentially less side effects than conventional medicine. For the pharmaceutical companies, bioactive natural products constitute attractive drug leads, as they have been optimized in a long-term natural selection process for optimal interaction with biomolecules. To promote the ecological survival of plants, structures of secondary products have evolved to interact with molecular targets affecting the cells, tissues and physiological functions in competing microorganisms, plants, and animals. In this, respect, some plant secondary products may exert their action by resembling endogenous metabolites, ligands, hormones, signal transduction molecules, or neurotransmitters and thus have beneficial effects on humans.
Future Perspectives
Metabolomics, the study of metabolism at the global level, is moving to exciting directions.With the development ofmore sensitive and advanced instrumentation and computational tools for data interpretation in the physiological context, metabolomics have the potential to impact our understanding of molecular mechanisms of diseases. A state-of-theart metabolomics study requires knowledge in many areas and especially at the interface of chemistry, biology, and
computer science. High-quality samples, improvements in automated metabolite identification, complete coverage of the human metabolome, establishment of spectral databases of metabolites and associated biochemical identities, innovative experimental designs to best address a hypothesis, as well as novel computational tools to handle metabolomics data are critical hurdles that must be overcome to drive the inclusion of metabolomics in all steps of drug discovery and drug development. The examples presented above demonstrated that metabolite profiles reflect both environmental and genetic influences in patients and reveal new links between metabolites and diseases providing needed prognostic,diagnostic, and surrogate biomarkers. The integration of these signatures with other omic technologies is of utmost importance to characterize the entire spectrum of malignant phenotype.
Volume Summary & Epilogue
Metabolomics Summary and Perspective
Author and Curator: Larry H Bernstein, MD, FCAP
This Summary & Epilogue has EIGHT parts, as follows:
Part 1
Metabolomics Continues Auspicious Climb
Part 2
Biologists Find ‘Missing Link’ in the Production of Protein Factories in Cells
Part 3
Neuroscience
Part 4
Cancer Research
Part 5
Metabolic Syndrome
Part 6
Biomarkers
Part 7
Epigenetics and Drug Metabolism
Part 8
Pictorial
This is the final article in a robust series on metabolism, metabolomics, and the “-OMICS-“ biological synthesis that is creating a more holistic and interoperable view of natural sciences, including the biological disciplines, climate science, physics, chemistry, toxicology, pharmacology, and pathophysiology with as yet unforeseen consequences.
There have been impressive advances already in the research into developmental biology, plant sciences, microbiology, mycology, and human diseases, most notably, cancer, metabolic , and infectious, as well as neurodegenerative diseases.



Part 1. MetabolomicsSurge

Part 1
Metabolomics Continues Auspicious Climb
Jeffery Herman, Ph.D.
GEN May 1, 2012 (Vol. 32, No. 9)
Aberrant biochemical and metabolite signaling plays an important role in
- the development and progression of diseased tissue.
This concept has been studied by the science community for decades. However, with relatively
- recent advances in analytical technology and bioinformatics as well as
- the development of the Human Metabolome Database (HMDB),
metabolomics has become an invaluable field of research.
At the “International Conference and Exhibition on Metabolomics & Systems Biology” held recently in San Francisco, researchers and industry leaders discussed how
- the underlying cellular biochemical/metabolite fingerprint in response to
- a specific disease state,
- toxin exposure, or
- pharmaceutical compound
- is useful in clinical diagnosis and biomarker discovery and
- in understanding disease development and progression.
Developed by BASF, MetaMap® Tox is
- a database that helps identify in vivo systemic effects of a tested compound, including
- targeted organs,
- mechanism of action, and
- adverse events.
Based on 28-day systemic rat toxicity studies, MetaMap Tox is composed of
- differential plasma metabolite profiles of rats
- after exposure to a large variety of chemical toxins and pharmaceutical compounds.
“Using the reference data,
- we have developed more than 110 patterns of metabolite changes, which are
- specific and predictive for certain toxicological modes of action,”
said Hennicke Kamp, Ph.D., group leader, department of experimental toxicology and ecology at BASF.
With MetaMap Tox, a potential drug candidate
- can be compared to a similar reference compound
- using statistical correlation algorithms,
- which allow for the creation of a toxicity and mechanism of action profile.
“MetaMap Tox, in the context of early pre-clinical safety enablement in pharmaceutical development,” continued Dr. Kamp,
- has been independently validated “
- by an industry consortium (Drug Safety Executive Council) of 12 leading biopharmaceutical companies.”
Dr. Kamp added that this technology may prove invaluable
- allowing for quick and accurate decisions and
- for high-throughput drug candidate screening, in evaluation
- on the safety and efficacy of compounds
- during early and preclinical toxicological studies,
- by comparing a lead compound to a variety of molecular derivatives, and
- the rapid identification of the most optimal molecular structure
- with the best efficacy and safety profiles might be streamlined.

Targeted Tandem Mass Spectrometry
Biocrates Life Sciences focuses on targeted metabolomics, an important approach for
- the accurate quantification of known metabolites within a biological sample.
Originally used for the clinical screening of inherent metabolic disorders from dried blood-spots of newborn children, Biocrates has developed
- a tandem mass spectrometry (MS/MS) platform, which allows for
- the identification,
- quantification, and
- mapping of more than 800 metabolites to specific cellular pathways.
It is based on flow injection analysis and high-performance liquid chromatography MS/MS.


The MetaDisIDQ® Kit is a
- “multiparamatic” diagnostic assay designed for the “comprehensive assessment of a person’s metabolic state” and
- the early determination of pathophysiological events with regards to a specific disease.
MetaDisIDQ is designed to quantify
- a diverse range of 181 metabolites involved in major metabolic pathways
- from a small amount of human serum (10 µL) using isotopically labeled internal standards,
This kit has been demonstrated to detect changes in metabolites that are commonly associated with the development of
- metabolic syndrome, type 2 diabetes, and diabetic nephropathy,
Dr. Dallman reports that data generated with the MetaDisIDQ kit correlates strongly with
- routine chemical analyses of common metabolites including glucose and creatinine
Biocrates has also developed the MS/MS-based AbsoluteIDQ® kits, which are
- an “easy-to-use” biomarker analysis tool for laboratory research.
The kit functions on MS machines from a variety of vendors, and allows for the quantification of 150-180 metabolites.
The SteroIDQ® kit is a high-throughput standardized MS/MS diagnostic assay,
- validated in human serum, for the rapid and accurate clinical determination of 16 known steroids.
Initially focusing on the analysis of steroid ranges for use in hormone replacement therapy, the SteroIDQ Kit is expected to have a wide clinical application.
Hormone-Resistant Breast Cancer
Scientists at Georgetown University have shown that
- breast cancer cells can functionally coordinate cell-survival and cell-proliferation mechanisms,
- while maintaining a certain degree of cellular metabolism.
To grow, cells need energy, and energy is a product of cellular metabolism. For nearly a century, it was thought that
- the uncoupling of glycolysis from the mitochondria,
- leading to the inefficient but rapid metabolism of glucose and
- the formation of lactic acid (the Warburg effect), was
the major and only metabolism driving force for unchecked proliferation and tumorigenesis of cancer cells.
Other aspects of metabolism were often overlooked.
“.. we understand now that
- cellular metabolism is a lot more than just metabolizing glucose,”
said Robert Clarke, Ph.D., professor of oncology and physiology and biophysics at Georgetown University. Dr. Clarke, in collaboration with the Waters Center for Innovation at Georgetown University (led by Albert J. Fornace, Jr., M.D.), obtained
- the metabolomic profile of hormone-sensitive and -resistant breast cancer cells through the use of UPLC-MS.
They demonstrated that breast cancer cells, through a rather complex and not yet completely understood process,
- can functionally coordinate cell-survival and cell-proliferation mechanisms,
- while maintaining a certain degree of cellular metabolism.
This is at least partly accomplished through the upregulation of important pro-survival mechanisms; including
- the unfolded protein response;
- a regulator of endoplasmic reticulum stress and
- initiator of autophagy.
Normally, during a stressful situation, a cell may
- enter a state of quiescence and undergo autophagy,
- a process by which a cell can recycle organelles
- in order to maintain enough energy to survive during a stressful situation or,
if the stress is too great,
- undergo apoptosis.
By integrating cell-survival mechanisms and cellular metabolism
- advanced ER+ hormone-resistant breast cancer cells
- can maintain a low level of autophagy
- to adapt and resist hormone/chemotherapy treatment.
This adaptation allows cells
- to reallocate important metabolites recovered from organelle degradation and
- provide enough energy to also promote proliferation.
With further research, we can gain a better understanding of the underlying causes of hormone-resistant breast cancer, with
- the overall goal of developing effective diagnostic, prognostic, and therapeutic tools.
NMR
Over the last two decades, NMR has established itself as a major tool for metabolomics analysis. It is especially adept at testing biological fluids. [Bruker BioSpin]
Historically, nuclear magnetic resonance spectroscopy (NMR) has been used forstructural elucidation of pure molecular compounds. However, in the last two decades, NMR has established itself as a major tool for metabolomics analysis. Since
- the integral of an NMR signal is directly proportional to
- the molar concentration throughout the dynamic range of a sample,
“the simultaneous quantification of compounds is possible
- without the need for specific reference standards or calibration curves,” according to Lea Heintz of Bruker BioSpin.
NMR is adept at testing biological fluids because of
- high reproducibility,
- standardized protocols,
- low sample manipulation, and
- the production of a large subset of data,
Bruker BioSpin is presently involved in a project for the screening of inborn errors of metabolism in newborn children from Turkey, based on their urine NMR profiles. More than 20 clinics are participating to the project that is coordinated by INFAI, a specialist in the transfer of advanced analytical technology into medical diagnostics. The construction of statistical models are being developed
- for the detection of deviations from normality, as well as
- automatic quantification methods for indicative metabolites
Bruker BioSpin recently installed high-resolution magic angle spinning NMR (HRMAS-NMR) systems that can rapidly analyze tissue biopsies. The main objective for HRMAS-NMR is to establish a rapid and effective clinical method to assess tumor grade and other important aspects of cancer during surgery.
Combined NMR and Mass Spec
There is increasing interest in combining NMR and MS, two of the main analytical assays in metabolomic research, as a means
- to improve data sensitivity and to
- fully elucidate the complex metabolome within a given biological sample.
- to realize a potential for cancer biomarker discovery in the realms of diagnosis, prognosis, and treatment.
Using combined NMR and MS to measure the levels of nearly 250 separate metabolites in the patient’s blood, Dr. Weljie and other researchers at the University of Calgary were able to rapidly determine the malignancy of a pancreatic lesion (in 10–15% of the cases, it is difficult to discern between benign and malignant), while avoiding unnecessary surgery in patients with benign lesions.
When performing NMR and MS on a single biological fluid, ultimately “we are,” noted Dr. Weljie,
- “splitting up information content, processing, and introducing a lot of background noise and error and
- then trying to reintegrate the data…
It’s like taking a complex item, with multiple pieces, out of an IKEA box and trying to repackage it perfectly into another box.”
By improving the workflow between the initial splitting of the sample, they improved endpoint data integration, proving that
- a streamlined approach to combined NMR/MS can be achieved,
- leading to a very strong, robust and precise metabolomics toolset.
Metabolomics Research Picks Up Speed
Field Advances in Quest to Improve Disease Diagnosis and Predict Drug Response
John Morrow Jr., Ph.D.
GEN May 1, 2011 (Vol. 31, No. 9)
As an important discipline within systems biology, metabolomics is being explored by a number of laboratories for
- its potential in pharmaceutical development.
Studying metabolites can offer insights into the relationships between genotype and phenotype, as well as between genotype and environment. In addition, there is plenty to work with—there are estimated to be some 2,900 detectable metabolites in the human body, of which
- 309 have been identified in cerebrospinal fluid,
- 1,122 in serum,
- 458 in urine, and
- roughly 300 in other compartments.
Guowang Xu, Ph.D., a researcher at the Dalian Institute of Chemical Physics. is investigating the causes of death in China,
- and how they have been changing over the years as the country has become a more industrialized nation.
- the increase in the incidence of metabolic disorders such as diabetes has grown to affect 9.7% of the Chinese population.
Dr. Xu, collaborating with Rainer Lehman, Ph.D., of the University of Tübingen, Germany, compared urinary metabolites in samples from healthy individuals with samples taken from prediabetic, insulin-resistant subjects. Using mass spectrometry coupled with electrospray ionization in the positive mode, they observed striking dissimilarities in levels of various metabolites in the two groups.
“When we performed a comprehensive two-dimensional gas chromatography, time-of-flight mass spectrometry analysis of our samples, we observed several metabolites, including
- 2-hydroxybutyric acid in plasma,
- as potential diabetes biomarkers,” Dr. Xu explains.
In other, unrelated studies, Dr. Xu and the German researchers used a metabolomics approach to investigate the changes in plasma metabolite profiles immediately after exercise and following a 3-hour and 24-hour period of recovery. They found that
- medium-chain acylcarnitines were the most distinctive exercise biomarkers, and
- they are released as intermediates of partial beta oxidation in human myotubes and mouse muscle tissue.
Dr. Xu says. “The traditional approach of assessment based on a singular biomarker is being superseded by the introduction of multiple marker profiles.”
Typical of the studies under way by Dr. Kaddurah-Daouk and her colleaguesat Duke University
- is a recently published investigation highlighting the role of an SNP variant in
- the glycine dehydrogenase gene on individual response to antidepressants.
- patients who do not respond to the selective serotonin uptake inhibitors citalopram and escitalopram
- carried a particular single nucleotide polymorphism in the GD gene.
“These results allow us to pinpoint a possible
- role for glycine in selective serotonin reuptake inhibitor response and
- illustrate the use of pharmacometabolomics to inform pharmacogenomics.
These discoveries give us the tools for prognostics and diagnostics so that
- we can predict what conditions will respond to treatment.
“This approach to defining health or disease in terms of metabolic states opens a whole new paradigm.
By screening hundreds of thousands of molecules, we can understand
- the relationship between human genetic variability and the metabolome.”
Dr. Kaddurah-Daouk talks about statins as a current
- model of metabolomics investigations.
It is now known that the statins have widespread effects, altering a range of metabolites. To sort out these changes and develop recommendations for which individuals should be receiving statins will require substantial investments of energy and resources into defining the complex web of biochemical changes that these drugs initiate.
Furthermore, Dr. Kaddurah-Daouk asserts that,
- “genetics only encodes part of the phenotypic response.
One needs to take into account the
- net environment contribution in order to determine
- how both factors guide the changes in our metabolic state that determine the phenotype.”
Interactive Metabolomics
Researchers at the University of Nottingham use diffusion-edited nuclear magnetic resonance spectroscopy to assess the effects of a biological matrix on metabolites. Diffusion-edited NMR experiments provide a way to
- separate the different compounds in a mixture
- based on the differing translational diffusion coefficients (which reflect the size and shape of the molecule).
The measurements are carried out by observing
- the attenuation of the NMR signals during a pulsed field gradient experiment.
Clare Daykin, Ph.D., is a lecturer at the University of Nottingham, U.K. Her field of investigation encompasses “interactive metabolomics,”which she defines as
“the study of the interactions between low molecular weight biochemicals and macromolecules in biological samples ..
- without preselection of the components of interest.
“Blood plasma is a heterogeneous mixture of molecules that
- undergo a variety of interactions including metal complexation,
- chemical exchange processes,
- micellar compartmentation,
- enzyme-mediated biotransformations, and
- small molecule–macromolecular binding.”
Many low molecular weight compounds can exist
- freely in solution,
- bound to proteins, or
- within organized aggregates such as lipoprotein complexes.
Therefore, quantitative comparison of plasma composition from
- diseased individuals compared to matched controls provides an incomplete insight to plasma metabolism.
“It is not simply the concentrations of metabolites that must be investigated,
- but their interactions with the proteins and lipoproteins within this complex web.
Rather than targeting specific metabolites of interest, Dr. Daykin’s metabolite–protein binding studies aim to study
- the interactions of all detectable metabolites within the macromolecular sample.
Such activities can be studied through the use of diffusion-edited nuclear magnetic resonance (NMR) spectroscopy, in which one can assess
- the effects of the biological matrix on the metabolites.
“This can lead to a more relevant and exact interpretation
- for systems where metabolite–macromolecule interactions occur.”
Diffusion-edited NMR experiments provide a way to separate the different compounds in a mixture based on
- the differing translational diffusion coefficients (which reflect the size and shape of the molecule).
The measurements are carried out by observing
- the attenuation of the NMR signals during a pulsed field gradient experiment.
Pushing the Limits
It is widely recognized that many drug candidates fail during development due to ancillary toxicity. Uwe Sauer, Ph.D., professor, and Nicola Zamboni, Ph.D., researcher, both at the Eidgenössische Technische Hochschule, Zürich (ETH Zürich), are applying
- high-throughput intracellular metabolomics to understand
- the basis of these unfortunate events and
- head them off early in the course of drug discovery.
“Since metabolism is at the core of drug toxicity, we developed a platform for
- measurement of 50–100 targeted metabolites by
- a high-throughput system consisting of flow injection
- coupled to tandem mass spectrometry.”
Using this approach, Dr. Sauer’s team focused on
- the central metabolism of the yeast Saccharomyces cerevisiae, reasoning that
- this core network would be most susceptible to potential drug toxicity.
Screening approximately 41 drugs that were administered at seven concentrations over three orders of magnitude, they observed changes in metabolome patterns at much lower drug concentrations without attendant physiological toxicity.
The group carried out statistical modeling of about
- 60 metabolite profiles for each drug they evaluated.
This data allowed the construction of a “profile effect map” in which
- the influence of each drug on metabolite levels can be followed, including off-target effects, which
- provide an indirect measure of the possible side effects of the various drugs.
Dr. Sauer says.“We have found that this approach is
- at least 100 times as fast as other omics screening platforms,”
“Some drugs, including many anticancer agents,
- disrupt metabolism long before affecting growth.”

Furthermore, they used the principle of 13C-based flux analysis, in which
- metabolites labeled with 13C are used to follow the utilization of metabolic pathways in the cell.
These 13C-determined intracellular responses of metabolic fluxes to drug treatment demonstrate
- the functional performance of the network to be rather robust,

leading Dr. Sauer to the conclusion that
- the phenotypic vigor he observes to drug challenges
- is achieved by a flexible make up of the metabolome.
Dr. Sauer is confident that it will be possible to expand the scope of these investigations to hundreds of thousands of samples per study. This will allow answers to the questions of
- how cells establish a stable functioning network in the face of inevitable concentration fluctuations.
Is Now the Hour?
There is great enthusiasm and agitation within the biotech community for
- metabolomics approaches as a means of reversing the dismal record of drug discovery
that has accumulated in the last decade.
While the concept clearly makes sense and is being widely applied today, there are many reasons why drugs fail in development, and metabolomics will not be a panacea for resolving all of these questions. It is too early at this point to recognize a trend or a track record, and it will take some time to see how this approach can aid in drug discovery and shorten the timeline for the introduction of new pharmaceutical agents.


Part 2
Biologists Find ‘Missing Link’ in the Production of Protein Factories in Cells
Biologists at UC San Diego have found
- the “missing link” in the chemical system that
- enables animal cells to produce ribosomes
—the thousands of protein “factories” contained within each cell that
- manufacture all of the proteins needed to build tissue and sustain life.

Their discovery, detailed in the June 23 issue of the journal Genes & Development, will not only force
- a revision of basic textbooks on molecular biology, but also
- provide scientists with a better understanding of
- how to limit uncontrolled cell growth, such as cancer,
- that might be regulated by controlling the output of ribosomes.
Ribosomes are responsible for the production of the wide variety of proteins that include
- enzymes;
- structural molecules, such as hair,
- skin and bones;
- hormones like insulin; and
- components of our immune system such as antibodies.
Regarded as life’s most important molecular machine, ribosomes have been intensively studied by scientists (the 2009 Nobel Prize in Chemistry, for example, was awarded for studies of its structure and function). But until now researchers had not uncovered all of the details of how the proteins that are used to construct ribosomes are themselves produced.
In multicellular animals such as humans,
- ribosomes are made up of about 80 different proteins
(humans have 79 while some other animals have a slightly different number) as well as - four different kinds of RNA molecules.
In 1969, scientists discovered that
- the synthesis of the ribosomal RNAs is carried out by specialized systems using two key enzymes:
- RNA polymerase I and RNA polymerase III.
But until now, scientists were unsure if a complementary system was also responsible for
- the production of the 80 proteins that make up the ribosome.
That’s essentially what the UC San Diego researchers headed by Jim Kadonaga, a professor of biology, set out to examine. What they found was the missing link—the specialized
- system that allows ribosomal proteins themselves to be synthesized by the cell.
Kadonaga says that he and coworkers found that ribosomal proteins are synthesized via
- a novel regulatory system with the enzyme RNA polymerase II and
- a factor termed TRF2,”
“For the production of most proteins,
- RNA polymerase II functions with
- a factor termed TBP,
- but for the synthesis of ribosomal proteins, it uses TRF2.”
- this specialized TRF2-based system for ribosome biogenesis
- provides a new avenue for the study of ribosomes and
- its control of cell growth, and
“it should lead to a better understanding and potential treatment of diseases such as cancer.”


Other authors of the paper were UC San Diego biologists Yuan-Liang Wang, Sascha Duttke and George Kassavetis, and Kai Chen, Jeff Johnston, and Julia Zeitlinger of the Stowers Institute for Medical Research in Kansas City, Missouri. Their research was supported by two grants from the National Institutes of Health (1DP2OD004561-01 and R01 GM041249).
Turning Off a Powerful Cancer Protein
Scientists have discovered how to shut down a master regulatory transcription factor that is
- key to the survival of a majority of aggressive lymphomas,
- which arise from the B cells of the immune system.
The protein, Bcl6, has long been considered too complex to target with a drug since it is also crucial
- to the healthy functioning of many immune cells in the body, not just B cells gone bad.
The researchers at Weill Cornell Medical College report that it is possible
- to shut down Bcl6 in diffuse large B-cell lymphoma (DLBCL)
- while not affecting its vital function in T cells and macrophages
- that are needed to support a healthy immune system.
If Bcl6 is completely inhibited, patients might suffer from systemic inflammation and atherosclerosis. The team conducted this new study to help clarify possible risks, as well as to understand
- how Bcl6 controls the various aspects of the immune system.
The findings in this study were inspired from
- preclinical testing of two Bcl6-targeting agents that Dr. Melnick and his Weill Cornell colleagues have developed
- to treat DLBCLs.
These experimental drugs are
- RI-BPI, a peptide mimic, and
- the small molecule agent 79-6.
“This means the drugs we have developed against Bcl6 are more likely to be
- significantly less toxic and safer for patients with this cancer than we realized,”
says Ari Melnick, M.D., professor of hematology/oncology and a hematologist-oncologist at NewYork-Presbyterian Hospital/Weill Cornell Medical Center.
Dr. Melnick says the discovery that
- a master regulatory transcription factor can be targeted
- offers implications beyond just treating DLBCL.
Recent studies from Dr. Melnick and others have revealed that
- Bcl6 plays a key role in the most aggressive forms of acute leukemia, as well as certain solid tumors.
Bcl6 can control the type of immune cell that develops in the bone marrow—playing many roles
- in the development of B cells, T cells, macrophages, and other cells—including a primary and essential role in
- enabling B-cells to generate specific antibodies against pathogens.
According to Dr. Melnick, “When cells lose control of Bcl6,
- lymphomas develop in the immune system.
Lymphomas are ‘addicted’ to Bcl6, and therefore
- Bcl6 inhibitors powerfully and quickly destroy lymphoma cells,” .
The big surprise in the current study is that rather than functioning as a single molecular machine,
- Bcl6 functions like a Swiss Army knife,
- using different tools to control different cell types.
This multifunction paradigm could represent a general model for the functioning of other master regulatory transcription factors.
“In this analogy, the Swiss Army knife, or transcription factor, keeps most of its tools folded,
- opening only the one it needs in any given cell type,”
He makes the following analogy:
- “For B cells, it might open and use the knife tool;
- for T cells, the cork screw;
- for macrophages, the scissors.”
“this means that you only need to prevent the master regulator from using certain tools to treat cancer. You don’t need to eliminate the whole knife,” . “In fact, we show that taking out the whole knife is harmful since
- the transcription factor has many other vital functions that other cells in the body need.”
Prior to these study results, it was not known that a master regulator could separate its functions so precisely. Researchers hope this will be a major benefit to the treatment of DLBCL and perhaps other disorders that are influenced by Bcl6 and other master regulatory transcription factors.
The study is published in the journal Nature Immunology, in a paper titled “Lineage-specific functions of Bcl-6 in immunity and inflammation are mediated by distinct biochemical mechanisms”.
Part 3
Neuroscience
Vesicles influence function of nerve cells
Oct, 06 2014 source: http://feeds.sciencedaily.com

Neurons (blue) which have absorbed exosomes (green) have increased levels of the enzyme catalase (red), which helps protect them against peroxides.
Tiny vesicles containing protective substances
- which they transmit to nerve cells apparently
- play an important role in the functioning of neurons.
As cell biologists at Johannes Gutenberg University Mainz (JGU) have discovered,
- nerve cells can enlist the aid of mini-vesicles of neighboring glial cells
- to defend themselves against stress and other potentially detrimental factors.
These vesicles, called exosomes, appear to stimulate the neurons on various levels:
- they influence electrical stimulus conduction,
- biochemical signal transfer, and
- gene regulation.
Exosomes are thus multifunctional signal emitters
- that can have a significant effect in the brain.

The researchers in Mainz already observed in a previous study that
- oligodendrocytes release exosomes on exposure to neuronal stimuli.
- these are absorbed by the neurons and improve neuronal stress tolerance.
Oligodendrocytes, a type of glial cell, form an
- insulating myelin sheath around the axons of neurons.
The exosomes transport protective proteins such as
- heat shock proteins,
- glycolytic enzymes, and
- enzymes that reduce oxidative stress from one cell type to another,
- but also transmit genetic information in the form of ribonucleic acids.
“As we have now discovered in cell cultures, exosomes seem to have a whole range of functions,” explained Dr. Eva-Maria Krmer-Albers. By means of their transmission activity, the small bubbles that are the vesicles
- not only promote electrical activity in the nerve cells, but also
- influence them on the biochemical and gene regulatory level.
“The extent of activities of the exosomes is impressive,” added Krmer-Albers. The researchers hope that the understanding of these processes will contribute to the development of new strategies for the treatment of neuronal diseases. Their next aim is to uncover how vesicles actually function in the brains of living organisms.
http://labroots.com/user/news/article/id/217438/title/vesicles-influence-function-of-nerve-cells
The above story is based on materials provided by Universitt Mainz.
Universitt Mainz. “Vesicles influence function of nerve cells.” ScienceDaily. ScienceDaily, 6 October 2014.www.sciencedaily.com/releases/2014/10/141006174214.htm
Neuroscientists use snail research to help explain “chemo brain”
10/08/2014
It is estimated that as many as half of patients taking cancer drugs experience a decrease in mental sharpness. While there have been many theories, what causes “chemo brain” has eluded scientists.
In an effort to solve this mystery, neuroscientists at The University of Texas Health Science Center at Houston (UTHealth) conducted an experiment in an animal memory model and their results point to a possible explanation. Findings appeared in The Journal of Neuroscience.
In the study involving a sea snail that shares many of the same memory mechanisms as humans and a drug used to treat a variety of cancers, the scientists identified
- memory mechanisms blocked by the drug.
Then, they were able to counteract or
- unblock the mechanisms by administering another agent.
“Our research has implications in the care of people given to cognitive deficits following drug treatment for cancer,” said John H. “Jack” Byrne, Ph.D., senior author, holder of the June and Virgil Waggoner Chair and Chairman of the Department of Neurobiology and Anatomy at the UTHealth Medical School. “There is no satisfactory treatment at this time.”
Byrne’s laboratory is known for its use of a large snail called Aplysia californica to further the understanding of the biochemical signaling among nerve cells (neurons). The snails have large neurons that relay information much like those in humans.
When Byrne’s team compared cell cultures taken from normal snails to
- those administered a dose of a cancer drug called doxorubicin,
the investigators pinpointed a neuronal pathway
- that was no longer passing along information properly.
With the aid of an experimental drug,
- the scientists were able to reopen the pathway.
Unfortunately, this drug would not be appropriate for humans, Byrne said. “We want to identify other drugs that can rescue these memory mechanisms,” he added.
According the American Cancer Society, some of the distressing mental changes cancer patients experience may last a short time or go on for years.
Byrne’s UT Health research team includes co-lead authors Rong-Yu Liu, Ph.D., and Yili Zhang, Ph.D., as well as Brittany Coughlin and Leonard J. Cleary, Ph.D. All are affiliated with the W.M. Keck Center for the Neurobiology of Learning and Memory.
Byrne and Cleary also are on the faculty of The University of Texas Graduate School of Biomedical Sciences at Houston. Coughlin is a student at the school, which is jointly operated by UT Health and The University of Texas MD Anderson Cancer Center.
The study titled “Doxorubicin Attenuates Serotonin-Induced Long-Term Synaptic Facilitation by Phosphorylation of p38 Mitogen-Activated Protein Kinase” received support from National Institutes of Health grant (NS019895) and the Zilkha Family Discovery Fellowship.
Doxorubicin Attenuates Serotonin-Induced Long-Term Synaptic Facilitation by Phosphorylation of p38 Mitogen-Activated Protein Kinase
Source: Univ. of Texas Health Science Center at Houston
http://www.rdmag.com/news/2014/10/neuroscientists-use-snail-research-help-explain-E2_9_Cchemo-brain
Doxorubicin Attenuates Serotonin-Induced Long-Term Synaptic Facilitation by Phosphorylation of p38 Mitogen-Activated Protein Kinase
Rong-Yu Liu*, Yili Zhang*, Brittany L. Coughlin, Leonard J. Cleary, and John H. Byrne +Show Affiliations
The Journal of Neuroscience, 1 Oct 2014, 34(40): 13289-13300;
http://dx.doi.org:/10.1523/JNEUROSCI.0538-14.2014
Doxorubicin (DOX) is an anthracycline used widely for cancer chemotherapy. Its primary mode of action appears to be
- topoisomerase II inhibition, DNA cleavage, and free radical generation.
However, in non-neuronal cells, DOX also inhibits the expression of
- dual-specificity phosphatases (also referred to as MAPK phosphatases) and thereby
- inhibits the dephosphorylation of extracellular signal-regulated kinase (ERK) and
- p38 mitogen-activated protein kinase (p38 MAPK),
- two MAPK isoforms important for long-term memory (LTM) formation.
Activation of these kinases by DOX in neurons, if present,
- could have secondary effects on cognitive functions, such as learning and memory.
The present study used cultures of rat cortical neurons and sensory neurons (SNs) of Aplysia
- to examine the effects of DOX on levels of phosphorylated ERK (pERK) and
- phosphorylated p38 (p-p38) MAPK.
In addition, Aplysia neurons were used to examine the effects of DOX on
- long-term enhanced excitability, long-term synaptic facilitation (LTF), and
- long-term synaptic depression (LTD).
DOX treatment led to elevated levels of
- pERK and p-p38 MAPK in SNs and cortical neurons.
In addition, it increased phosphorylation of
- the downstream transcriptional repressor cAMP response element-binding protein 2 in SNs.
DOX treatment blocked serotonin-induced LTF and enhanced LTD induced by the neuropeptide Phe-Met-Arg-Phe-NH2. The block of LTF appeared to be attributable to
- overriding inhibitory effects of p-p38 MAPK, because
- LTF was rescued in the presence of an inhibitor of p38 MAPK
(SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole]) .
These results suggest that acute application of DOX might impair the formation of LTM via the p38 MAPK pathway.
Terms: Aplysia chemotherapy ERK p38 MAPK serotonin synaptic plasticity
Technology that controls brain cells with radio waves earns early BRAIN grant
10/08/2014

BRAIN control: The new technology uses radio waves to activate or silence cells remotely. The bright spots above represent cells with increased calcium after treatment with radio waves, a change that would allow neurons to fire.
A proposal to develop a new way to
- remotely control brain cells
from Sarah Stanley, a research associate in Rockefeller University’s Laboratory of Molecular Genetics, headed by Jeffrey M. Friedman, is
- among the first to receive funding from U.S. President Barack Obama’s BRAIN initiative.
The project will make use of a technique called
- radiogenetics that combines the use of radio waves or magnetic fields with
- nanoparticles to turn neurons on or off.
The National Institutes of Health is one of four federal agencies involved in the BRAIN (Brain Research through Advancing Innovative Neurotechnologies) initiative. Following in the ambitious footsteps of the Human Genome Project, the BRAIN initiative seeks
- to create a dynamic map of the brain in action,
a goal that requires the development of new technologies. The BRAIN initiative working group, which outlined the broad scope of the ambitious project, was co-chaired by Rockefeller’s Cori Bargmann, head of the Laboratory of Neural Circuits and Behavior.
Stanley’s grant, for $1.26 million over three years, is one of 58 projects to get BRAIN grants, the NIH announced. The NIH’s plan for its part of this national project, which has been pitched as “America’s next moonshot,” calls for $4.5 billion in federal funds over 12 years.
The technology Stanley is developing would
- enable researchers to manipulate the activity of neurons, as well as other cell types,
- in freely moving animals in order to better understand what these cells do.
Other techniques for controlling selected groups of neurons exist, but her newnanoparticle-based technique has a
- unique combination of features that may enable new types of experimentation.
- it would allow researchers to rapidly activate or silence neurons within a small area of the brain or
- dispersed across a larger region, including those in difficult-to-access locations.
Stanley also plans to explore the potential this method has for use treating patients.
“Francis Collins, director of the NIH, has discussed
- the need for studying the circuitry of the brain,
- which is formed by interconnected neurons.
Our remote-control technology may provide a tool with which researchers can ask new questions about the roles of complex circuits in regulating behavior,” Stanley says.
Rockefeller University’s Laboratory of Molecular Genetics
Source: Rockefeller Univ.
Part 4
Cancer Research
Two Proteins Found to Block Cancer Metastasis
Why do some cancers spread while others don’t? Scientists have now demonstrated that
- metastatic incompetent cancers actually “poison the soil”
- by generating a micro-environment that blocks cancer cells
- from settling and growing in distant organs.
The “seed and the soil” hypothesis proposed by Stephen Paget in 1889 is now widely accepted to explain how
- cancer cells (seeds) are able to generate fertile soil (the micro-environment)
- in distant organs that promotes cancer’s spread.
However, this concept had not explained why some tumors do not spread or metastasize.
The researchers, from Weill Cornell Medical College, found that
- two key proteins involved in this process work by
- dramatically suppressing cancer’s spread.
The study offers hope that a drug based on these
- potentially therapeutic proteins, prosaposin and Thrombospondin 1 (Tsp-1),
might help keep human cancer at bay and from metastasizing.
Scientists don’t understand why some tumors wouldn’t “want” to spread. It goes against their “job description,” says the study’s senior investigator, Vivek Mittal, Ph.D., an associate professor of cell and developmental biology in cardiothoracic surgery and director of the Neuberger Berman Foundation Lung Cancer Laboratory at Weill Cornell Medical College. He theorizes that metastasis occurs when
- the barriers that the body throws up to protect itself against cancer fail.
But there are some tumors in which some of the barriers may still be intact. “So that suggests
- those primary tumors will continue to grow, but that
- an innate protective barrier still exists that prevents them from spreading and invading other organs,”
The researchers found that, like typical tumors,
- metastasis-incompetent tumors also send out signaling molecules
- that establish what is known as the “premetastatic niche” in distant organs.
These niches composed of bone marrow cells and various growth factors have been described previously by others including Dr. Mittal as the fertile “soil” that the disseminated cancer cell “seeds” grow in.
Weill Cornell’s Raúl Catena, Ph.D., a postdoctoral fellow in Dr. Mittal’s laboratory, found an important difference between the tumor types.
Metastatic-incompetent tumors
- systemically increased expression of Tsp-1, a molecule known to fight cancer growth.
- increased Tsp-1 production was found specifically in the bone marrow myeloid cells
- that comprise the metastatic niche.
These results were striking, because for the first time Dr. Mittal says
- the bone marrow-derived myeloid cells were implicated as
- the main producers of Tsp-1,.
In addition, Weill Cornell and Harvard researchers found that
- prosaposin secreted predominantly by the metastatic-incompetent tumors
- increased expression of Tsp-1 in the premetastatic lungs.
Thus, Dr. Mittal posits that prosaposin works in combination with Tsp-1
- to convert pro-metastatic bone marrow myeloid cells in the niche
- into cells that are not hospitable to cancer cells that spread from a primary tumor.
- “The very same myeloid cells in the niche that we know can promote metastasis
- can also be induced under the command of the metastatic incompetent primary tumor to inhibit metastasis,”
The research team found that
- the Tsp-1–inducing activity of prosaposin
- was contained in only a 5-amino acid peptide region of the protein, and
- this peptide alone induced Tsp-1 in the bone marrow cells and
- effectively suppressed metastatic spread in the lungs
- in mouse models of breast and prostate cancer.
This 5-amino acid peptide with Tsp-1–inducing activity
- has the potential to be used as a therapeutic agent against metastatic cancer,
The scientists have begun to test prosaposin in other tumor types or metastatic sites.
Dr. Mittal says that “The clinical implications of the study are:
- “Not only is it theoretically possible to design a prosaposin-based drug or drugs
- that induce Tsp-1 to block cancer spread, but
- you could potentially create noninvasive prognostic tests
- to predict whether a cancer will metastasize.”
The study was reported in the April 30 issue of Cancer Discovery, in a paper titled “Bone Marrow-Derived Gr1+ Cells Can Generate a Metastasis-Resistant Microenvironment Via Induced Secretion of Thrombospondin-1”.
Disabling Enzyme Cripples Tumors, Cancer Cells

Published: Sep 05, 2013 http://www.technologynetworks.com/Metabolomics/news.aspx?id=157138
Knocking out a single enzyme dramatically cripples the ability of aggressive cancer cells to spread and grow tumors.
The paper, published in the journal Proceedings of the National Academy of Sciences, sheds new light on the importance of lipids, a group of molecules that includes fatty acids and cholesterol, in the development of cancer.
Researchers have long known that cancer cells metabolize lipids differently than normal cells. Levels of ether lipids – a class of lipids that are harder to break down – are particularly elevated in highly malignant tumors.
“Cancer cells make and use a lot of fat and lipids, and that makes sense because cancer cells divide and proliferate at an accelerated rate, and to do that,
- they need lipids, which make up the membranes of the cell,”
said study principal investigator Daniel Nomura, assistant professor in UC Berkeley’s Department of Nutritional Sciences and Toxicology. “Lipids have a variety of uses for cellular structure, but what we’re showing with our study is that
- lipids can send signals that fuel cancer growth.”
In the study, Nomura and his team tested the effects of reducing ether lipids on human skin cancer cells and primary breast tumors. They targeted an enzyme,
- alkylglycerone phosphate synthase, or AGPS,
- known to be critical to the formation of ether lipids.
The researchers confirmed that
- AGPS expression increased when normal cells turned cancerous.
- inactivating AGPS substantially reduced the aggressiveness of the cancer cells.
“The cancer cells were less able to move and invade,” said Nomura.
The researchers also compared the impact of
- disabling the AGPS enzyme in mice that had been injected with cancer cells.
Nomura. observes -“Among the mice that had the AGPS enzyme inactivated,
- the tumors were nonexistent,”
“The mice that did not have this enzyme
- disabled rapidly developed tumors.”
The researchers determined that
- inhibiting AGPS expression depleted the cancer cells of ether lipids.
- AGPS altered levels of other types of lipids important to the ability of the cancer cells to survive and spread, including
- prostaglandins and acyl phospholipids.
“What makes AGPS stand out as a treatment target is that the enzyme seems to simultaneously
- regulate multiple aspects of lipid metabolism
- important for tumor growth and malignancy.”
Future steps include the
- development of AGPS inhibitors for use in cancer therapy,
“This study sheds considerable light on the important role that AGPS plays in ether lipid metabolism in cancer cells, and it suggests that
- inhibitors of this enzyme could impair tumor formation,”
said Benjamin Cravatt, Professor and Chair of Chemical Physiology at The Scripps Research Institute, who is not part of the UC.
Agilent Technologies Thought Leader Award Supports Translational Research Program
Published: Mon, March 04, 2013
The award will support Dr DePinho’s research into
- metabolic reprogramming in the earliest stages of cancer.
Agilent Technologies Inc. announces that Dr. Ronald A. DePinho, a world-renowned oncologist and researcher, has received an Agilent Thought Leader Award.
DePinho is president of the University of Texas MD Anderson Cancer Center. DePinho and his team hope to discover and characterize
- alterations in metabolic flux during tumor initiation and maintenance, and to identify biomarkers for early detection of pancreatic cancer together with
- novel therapeutic targets.
Researchers on his team will work with scientists from the university’s newly formed Institute of Applied Cancer Sciences.
The Agilent Thought Leader Award provides funds to support personnel as well as a state-of-the-art Agilent 6550 iFunnel Q-TOF LC/MS system.
“I am extremely pleased to receive this award for metabolomics research, as the survival rates for pancreatic cancer have not significantly improved over the past 20 years,” DePinho said. “This technology will allow us to
- rapidly identify new targets that drive the formation, progression and maintenance of pancreatic cancer.
Discoveries from this research will also lead to
- the development of effective early detection biomarkers and novel therapeutic interventions.”
“We are proud to support Dr. DePinho’s exciting translational research program, which will make use of
- metabolomics and integrated biology workflows and solutions in biomarker discovery,”
said Patrick Kaltenbach, Agilent vice president, general manager of the Liquid Phase Division, and the executive sponsor of this award.
The Agilent Thought Leader Program promotes fundamental scientific advances by support of influential thought leaders in the life sciences and chemical analysis fields.
The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis
Ping Xie, Minghua Zhang, Shan He, Kefeng Lu, Yuhan Chen, Guichun Xing, et al.
Nature Communications 2014; 5(3733). http://dx.doi.org:/10.1038/ncomms4733
Neddylation, the covalent attachment of ubiquitin-like protein Nedd8, of the Cullin-RING E3 ligase family
- regulates their ubiquitylation activity.
However, regulation of HECT ligases by neddylation has not been reported to date. Here we show that
- the C2-WW-HECT ligase Smurf1 is activated by neddylation.
Smurf1 physically interacts with
- Nedd8 and Ubc12,
- forms a Nedd8-thioester intermediate, and then
- catalyses its own neddylation on multiple lysine residues.
Intriguingly, this autoneddylation needs
- an active site at C426 in the HECT N-lobe.
Neddylation of Smurf1 potently enhances
- ubiquitin E2 recruitment and
- augments the ubiquitin ligase activity of Smurf1.
The regulatory role of neddylation
- is conserved in human Smurf1 and yeast Rsp5.
Furthermore, in human colorectal cancers,
- the elevated expression of Smurf1, Nedd8, NAE1 and Ubc12
- correlates with cancer progression and poor prognosis.
These findings provide evidence that
- neddylation is important in HECT ubiquitin ligase activation and
- shed new light on the tumour-promoting role of Smurf1.

Subject terms: Biological sciences Cancer Cell biology
Figure 1: Smurf1 expression is elevated in colorectal cancer tissues.

(a) Smurf1 expression scores are shown as box plots, with the horizontal lines representing the median; the bottom and top of the boxes representing the 25th and 75th percentiles, respectively; and the vertical bars representing the ra
Figure 2: Positive correlation of Smurf1 expression with Nedd8 and its interacting enzymes in colorectal cancer.

(a) Representative images from immunohistochemical staining of Smurf1, Ubc12, NAE1 and Nedd8 in the same colorectal cancer tumour. Scale bars, 100 μm. (b–d) The expression scores of Nedd8 (b, n=283 ), NAE1 (c, n=281) and Ubc12 (d,n=19…
Figure 3: Smurf1 interacts with Ubc12.

(a) GST pull-down assay of Smurf1 with Ubc12. Both input and pull-down samples were subjected to immunoblotting with anti-His and anti-GST antibodies. Smurf1 interacted with Ubc12 and UbcH5c, but not with Ubc9. (b) Mapping the regions…
Figure 4: Nedd8 is attached to Smurf1through C426-catalysed autoneddylation.

(a) Covalent neddylation of Smurf1 in vitro.Purified His-Smurf1-WT or C699A proteins were incubated with Nedd8 and Nedd8-E1/E2. Reactions were performed as described in the Methods section. Samples were analysed by western blotting wi…
Figure 5: Neddylation of Smurf1 activates its ubiquitin ligase activity.

(a) In vivo Smurf1 ubiquitylation assay. Nedd8 was co-expressed with Smurf1 WT or C699A in HCT116 cells (left panels). Twenty-four hours post transfection, cells were treated with MG132 (20 μM, 8 h). HCT116 cells were transfected with…
http://www.nature.com/ncomms/2014/140513/ncomms4733/carousel/ncomms4733-f1.jpg
{kind=link}
http://www.nature.com/ncomms/2014/140513/ncomms4733/carousel/ncomms4733-f2.jpg
{kind=link}
http://www.nature.com/ncomms/2014/140513/ncomms4733/carousel/ncomms4733-f3.jpg
{kind=link}
http://www.nature.com/ncomms/2014/140513/ncomms4733/carousel/ncomms4733-f4.jpg
{kind=link}
http://www.nature.com/ncomms/2014/140513/ncomms4733/carousel/ncomms4733-f5.jpg
{kind=link}
http://www.nature.com/ncomms/2014/140513/ncomms4733/carousel/ncomms4733-f6.jpg
{kind=link}
http://www.nature.com/ncomms/2014/140513/ncomms4733/carousel/ncomms4733-f7.jpg
{kind=link}
http://www.nature.com/ncomms/2014/140513/ncomms4733/carousel/ncomms4733-f8.jpg
{kind=link}
The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response
M Simicek, S Lievens, M Laga, D Guzenko, VN. Aushev, et al.
Nature Cell Biology 2013; 15, 1220–1230 http://dx.doi.org:/10.1038/ncb2847
The RAS-like GTPase RALB mediates cellular responses to nutrient availability or viral infection by respectively
- engaging two components of the exocyst complex, EXO84 and SEC5.
- RALB employs SEC5 to trigger innate immunity signalling, whereas
- RALB–EXO84 interaction induces autophagocytosis.
How this differential interaction is achieved molecularly by the RAL GTPase remains unknown.
We found that whereas GTP binding
- turns on RALB activity,
ubiquitylation of RALB at Lys 47
- tunes its activity towards a particular effector.
Specifically, ubiquitylation at Lys 47
- sterically inhibits RALB binding to EXO84, while
- facilitating its interaction with SEC5.
Double-stranded RNA promotes
- RALB ubiquitylation and
- SEC5–TBK1 complex formation.
In contrast, nutrient starvation
- induces RALB deubiquitylation
- by accumulation and relocalization of the deubiquitylase USP33
- to RALB-positive vesicles.
Deubiquitylated RALB
- promotes the assembly of the RALB–EXO84–beclin-1 complexes
- driving autophagosome formation. Thus,
- ubiquitylation within the effector-binding domain
- provides the switch for the dual functions of RALB in
- autophagy and innate immune responses.
Part 5
Metabolic Syndrome
Single Enzyme is Necessary for Development of Diabetes
Published: Aug 20, 2014 http://www.technologynetworks.com/Metabolomics/news.aspx?ID=169416
12-LO enzyme promotes the obesity-induced oxidative stress in the pancreatic cells.
An enzyme called 12-LO promotes the obesity-induced oxidative stress in the pancreatic cells that leads
- to pre-diabetes, and diabetes.
12-LO’s enzymatic action is the last step in
- the production of certain small molecules that harm the cell,
according to a team from Indiana University School of Medicine, Indianapolis.
The findings will enable the development of drugs that can interfere with this enzyme, preventing or even reversing diabetes. The research is published ahead of print in the journal Molecular and Cellular Biology.
In earlier studies, these researchers and their collaborators at Eastern Virginia Medical School showed that
- 12-LO (which stands for 12-lipoxygenase) is present in these cells
- only in people who become overweight.
The harmful small molecules resulting from 12-LO’s enzymatic action are known asHETEs, short for hydroxyeicosatetraenoic acid.
- HETEs harm the mitochondria, which then
- fail to produce sufficient energy to enable
- the pancreatic cells to manufacture the necessary quantities of insulin.
For the study, the investigators genetically engineered mice that
- lacked the gene for 12-LO exclusively in their pancreas cells.
Mice were either fed a low-fat or high-fat diet.
Both the control mice and the knockout mice on the high fat diet
- developed obesity and insulin resistance.
The investigators also examined the pancreatic beta cells of both knockout and control mice, using both microscopic studies and molecular analysis. Those from the knockout mice were intact and healthy, while
- those from the control mice showed oxidative damage,
- demonstrating that 12-LO and the resulting HETEs
- caused the beta cell failure.
Mirmira notes that fatty diet used in the study was the Western Diet, which comprises mostly saturated-“bad”-fats. Based partly on a recent study of related metabolic pathways, he says that
- the unsaturated and mono-unsaturated fats-which comprise most fats in the healthy,
- relatively high fat Mediterranean diet-are unlikely to have the same effects.
“Our research is the first to show that 12-LO in the beta cell
- is the culprit in the development of pre-diabetes, following high fat diets,” says Mirmira.
“Our work also lends important credence to the notion that
- the beta cell is the primary defective cell in virtually all forms of diabetes and pre-diabetes.”
A New Player in Lipid Metabolism Discovered
Published: Aug18, 2014 http://www.technologynetworks.com/Metabolomics/news.aspx?ID=169356
Specially engineered mice gained no weight, and normal counterparts became obese
- on the same high-fat, obesity-inducing Western diet.
Specially engineered mice that lacked a particular gene did not gain weight
- when fed a typical high-fat, obesity-inducing Western diet.
Yet, these mice ate the same amount as their normal counterparts that became obese.
The mice were engineered with fat cells that lacked a gene called SEL1L,
- known to be involved in the clearance of mis-folded proteins
- in the cell’s protein making machinery called the endoplasmic reticulum (ER).
When mis-folded proteins are not cleared but accumulate,
- they destroy the cell and contribute to such diseases as
- mad cow disease,
- Type 1 diabetes and
- cystic fibrosis.
“The million-dollar question is why don’t these mice gain weight? Is this related to its inability to clear mis-folded proteins in the ER?” said Ling Qi, associate professor of molecular and biochemical nutrition and senior author of the study published online July 24 in Cell Metabolism. Haibo Sha, a research associate in Qi’s lab, is the paper’s lead author.
Interestingly, the experimental mice developed a host of other problems, including
- postprandial hypertriglyceridemia,
- and fatty livers.
“Although we are yet to find out whether these conditions contribute to the lean phenotype, we found that
- there was a lipid partitioning defect in the mice lacking SEL1L in fat cells,
- where fat cells cannot store fat [lipids], and consequently
- fat goes to the liver.
During the investigation of possible underlying mechanisms, we discovered
- a novel function for SEL1L as a regulator of lipid metabolism,” said Qi.
Sha said “We were very excited to find that
- SEL1L is required for the intracellular trafficking of
- lipoprotein lipase (LPL), acting as a chaperone,” .
and added that “Using several tissue-specific knockout mouse models,
- we showed that this is a general phenomenon,”
Without LPL, lipids remain in the circulation;
- fat and muscle cells cannot absorb fat molecules for storage and energy combustion,
People with LPL mutations develop
- postprandial hypertriglyceridemia similar to
- conditions found in fat cell-specific SEL1L-deficient mice, said Qi.
Future work will investigate the
- role of SEL1L in human patients carrying LPL mutations and
- determine why fat cell-specific SEL1L-deficient mice remain lean under Western diets, said Sha.
Co-authors include researchers from Cedars-Sinai Medical Center in Los Angeles; Wageningen University in the Netherlands; Georgia State University; University of California, Los Angeles; and the Medical College of Soochow University in China.
The study was funded by the U.S. National Institutes of Health, the Netherlands Organization for Health Research and Development National Institutes of Health, the Cedars-Sinai Medical Center, Chinese National Science Foundation, the American Diabetes Association, Cornell’s Center for Vertebrate Genomics and the Howard Hughes Medical Institute.
Part 6
Biomarkers
Biomarkers Take Center Stage
Josh P. Roberts
GEN May 1, 2013 (Vol. 33, No. 9) http://www.genengnews.com/
While work with biomarkers continues to grow, scientists are also grappling with research-related bottlenecks, such as
- affinity reagent development,
- platform reproducibility, and
- sensitivity.
Biomarkers by definition indicate some state or process that generally occurs
- at a spatial or temporal distance from the marker itself, and
it would not be an exaggeration to say that biomedicine has become infatuated with them:
- where to find them,
- when they may appear,
- what form they may take, and
- how they can be used to diagnose a condition or
- predict whether a therapy may be successful.
Biomarkers are on the agenda of many if not most industry gatherings, and in cases such as Oxford Global’s recent “Biomarker Congress” and the GTC “Biomarker Summit”, they hold the naming rights. There, some basic principles were built upon, amended, and sometimes challenged.
In oncology, for example, biomarker discovery is often predicated on the premise that
- proteins shed from a tumor will traverse to and persist in, and be detectable in, the circulation.
By quantifying these proteins—singularly or as part of a larger “signature”—the hope is
- to garner information about the molecular characteristics of the cancer
- that will help with cancer detection and
- personalization of the treatment strategy.
Yet this approach has not yet turned into the panacea that was hoped for. Bottlenecks exist in
- affinity reagent development,
- platform reproducibility, and
- sensitivity.
There is also a dearth of understanding of some of the
- fundamental principles of biomarker biology that we need to know the answers to,
said Parag Mallick, Ph.D., whose lab at Stanford University is “working on trying to understand where biomarkers come from.”
There are dogmas saying that
- circulating biomarkers come solely from secreted proteins.
But Dr. Mallick’s studies indicate that fully
- 50% of circulating proteins may come from intracellular sources or
- proteins that are annotated as such.
“We don’t understand the processes governing
- which tumor-derived proteins end up in the blood.”
Other questions include “how does the size of a tumor affect how much of a given protein will be in the blood?”—perhaps
- the tumor is necrotic at the center, or
- it’s hypervascular or hypovascular.
He points out “The problem is that these are highly nonlinear processes at work, and
- there is a large number of factors that might affect the answer to that question,” .
Their research focuses on using
- mass spectrometry and
- computational analysis
- to characterize the biophysical properties of the circulating proteome, and
- relate these to measurements made of the tumor itself.
Furthermore, he said – “We’ve observed that the proteins that are likely to
- first show up and persist in the circulation, ..
- are more stable than proteins that don’t,”
- “we can quantify how significant the effect is.”
The goal is ultimately to be able to
- build rigorous, formal mathematical models that will allow something measured in the blood
- to be tied back to the molecular biology taking place in the tumor.
And conversely, to use those models
- to predict from a tumor what will be found in the circulation.
“Ultimately, the models will allow you to connect the dots between
- what you measure in the blood and the biology of the tumor.”
Bound for Affinity Arrays
Affinity reagents are the main tools for large-scale protein biomarker discovery. And while this has tended to mean antibodies (or their derivatives), other affinity reagents are demanding a place in the toolbox.
Affimers, a type of affinity reagent being developed by Avacta, consist of
- a biologically inert, biophysically stable protein scaffold
- containing three variable regions into which
- distinct peptides are inserted.
The resulting three-dimensional surface formed by these peptides
- interacts and binds to proteins and other molecules in solution,
- much like the antigen-binding site of antibodies.
Unlike antibodies, Affimers are relatively small (13 KDa),
- non-post-translationally modified proteins
- that can readily be expressed in bacterial culture.
They may be made to bind surfaces through unique residues
- engineered onto the opposite face of the Affimer,
- allowing the binding site to be exposed to the target in solution.
“We don’t seem to see in what we’ve done so far
- any real loss of activity or functionality of Affimers when bound to surfaces—
they’re very robust,” said CEO Alastair Smith, Ph.D.
Avacta is taking advantage of this stability and its large libraries of Affimers to develop
- very large affinity microarrays for
- drug and biomarker discovery.
To date they have printed arrays with around 20–25,000 features, and Dr. Smith is “sure that we can get toward about 50,000 on a slide,” he said. “There’s no real impediment to us doing that other than us expressing the proteins and getting on with it.”
Customers will be provided with these large, complex “naïve” discovery arrays, readable with standard equipment. The plan is for the company to then “support our customers by providing smaller arrays with
- the Affimers that are binding targets of interest to them,” Dr. Smith foretold.
And since the intellectual property rights are unencumbered,
- Affimers in those arrays can be licensed to the end users
- to develop diagnostics that can be validated as time goes on.
Around 20,000-Affimer discovery arrays were recently tested by collaborator Professor Ann Morgan of the University of Leeds with pools of unfractionated serum from patients with symptoms of inflammatory disease. The arrays
- “rediscovered” elevated C-reactive protein (CRP, the clinical gold standard marker)
- as well as uncovered an additional 22 candidate biomarkers.
- other candidates combined with CRP, appear able to distinguish between different diseases such as
- rheumatoid arthritis,
- psoriatic arthritis,
- SLE, or
- giant cell arteritis.
Epigenetic Biomarkers

Sometimes biomarkers are used not to find disease but
- to distinguish healthy human cell types, with
- examples being found in flow cytometry and immunohistochemistry.
These widespread applications, however, are difficult to standardize, being
- subject to arbitrary or subjective gating protocols and other imprecise criteria.
Epiontis instead uses an epigenetic approach. “What we need is a unique marker that is
- demethylated only in one cell type and
- methylated in all the other cell types,”
Each cell of the right cell type will have
- two demethylated copies of a certain gene locus,
- allowing them to be enumerated by quantitative PCR.
The biggest challenge is finding that unique epigenetic marker. To do so they look through the literature for proteins and genes described as playing a role in the cell type’s biology, and then
- look at the methylation patterns to see if one can be used as a marker,
They also “use customized Affymetrix chips to look at the
- differential epigenetic status of different cell types on a genomewide scale.”
explained CBO and founder Ulrich Hoffmueller, Ph.D.
The company currently has a panel of 12 assays for 12 immune cell types. Among these is an assay for
- regulatory T (Treg) cells that queries the Foxp3 gene—which is uniquely demethylated in Treg
- even though it is transiently expressed in activated T cells of other subtypes.
Also assayed are Th17 cells, difficult to detect by flow cytometry because
- “the cells have to be stimulated in vitro,” he pointed out.
Developing New Assays for Cancer Biomarkers
Researchers at Myriad RBM and the Cancer Prevention Research Institute of Texas are collaborating to develop
- new assays for cancer biomarkers on the Myriad RBM Multi-Analyte Profile (MAP) platform.
The release of OncologyMAP 2.0 expanded Myriad RBM’s biomarker menu to over 250 analytes, which can be measured from a small single sample, according to the company. Using this menu, L. Stephen et al., published a poster, “Analysis of Protein Biomarkers in Prostate and Colorectal Tumor Lysates,” which showed the results of
- a survey of proteins relevant to colorectal (CRC) and prostate (PC) tumors
- to identify potential proteins of interest for cancer research.
The study looked at CRC and PC tumor lysates and found that 102 of the 115 proteins showed levels above the lower limit of quantification.
- Four markers were significantly higher in PC and 10 were greater in CRC.
For most of the analytes, duplicate sections of the tumor were similar, although some analytes did show differences. In four of the CRC analytes, tumor number four showed differences for CEA and tumor number 2 for uPA.
Thirty analytes were shown to be
- different in CRC tumor compared to its adjacent tissue.
- Ten of the analytes were higher in adjacent tissue compared to CRC.
- Eighteen of the markers examined demonstrated —-
significant correlations of CRC tumor concentration to serum levels.
“This suggests.. that the Oncology MAP 2.0 platform “provides a good method for studying changes in tumor levels because many proteins can be assessed with a very small sample.”
Clinical Test Development with MALDI-ToF
While there have been many attempts to translate results from early discovery work on the serum proteome into clinical practice, few of these efforts have progressed past the discovery phase.
Matrix-assisted laser desorption/ionization-time of flight (MALDI-ToF) mass spectrometry on unfractionated serum/plasma samples offers many practical advantages over alternative techniques, and does not require
- a shift from discovery to development and commercialization platforms.
Biodesix claims it has been able to develop the technology into
- a reproducible, high-throughput tool to
- routinely measure protein abundance from serum/plasma samples.
“.. we improved data-analysis algorithms to
- reproducibly obtain quantitative measurements of relative protein abundance from MALDI-ToF mass spectra.
Heinrich Röder, CTO points out that the MALDI-ToF measurements
- are combined with clinical outcome data using
- modern learning theory techniques
- to define specific disease states
- based on a patient’s serum protein content,”
The clinical utility of the identification of these disease states can be investigated through a retrospective analysis of differing sample sets. For example, Biodesix clinically validated its first commercialized serum proteomic test, VeriStrat®, in 85 different retrospective sample sets.
Röder adds that “It is becoming increasingly clear that
- the patients whose serum is characterized as VeriStrat Poor show
- consistently poor outcomes irrespective of
- tumor type,
- histology, or
- molecular tumor characteristics,”
MALDI-ToF mass spectrometry, in its standard implementation,
- allows for the observation of around 100 mostly high-abundant serum proteins.
Further, “while this does not limit the usefulness of tests developed from differential expression of these proteins,
- the discovery potential would be greatly enhanced
- if we could probe deeper into the proteome
- while not giving up the advantages of the MALDI-ToF approach,”
Biodesix reports that its new MALDI approach, Deep MALDI™, can perform
- simultaneous quantitative measurement of more than 1,000 serum protein features (or peaks) from 10 µL of serum in a high-throughput manner.
- it increases the observable signal noise ratio from a few hundred to over 50,000,
- resulting in the observation of many lower-abundance serum proteins.
Breast cancer, a disease now considered to be a collection of many complexes of symptoms and signatures—the dominant ones are labeled Luminal A, Luminal B, Her2, and Basal— which suggests different prognose, and
- these labels are considered too simplistic for understanding and managing a woman’s cancer.
Studies published in the past year have looked at
- somatic mutations,
- gene copy number aberrations,
- gene expression abnormalities,
- protein and miRNA expression, and
- DNA methylation,
coming up with a list of significantly mutated genes—hot spots—in different categories of breast cancers. Targeting these will inevitably be the focus of much coming research.
“We’ve been taking these large trials and profiling these on a variety of array or sequence platforms. We think we’ll get
- prognostic drivers
- predictive markers for taxanes and
- monoclonal antibodies and
- tamoxifen and aromatase inhibitors,”
explained Brian Leyland-Jones, Ph.D., director of Edith Sanford Breast Cancer Research. “We will end up with 20–40 different diseases, maybe more.”
Edith Sanford Breast Cancer Research is undertaking a pilot study in collaboration with The Scripps Research Institute, using a variety of tests on 25 patients to see how the information they provide complements each other, the overall flow, and the time required to get and compile results.
Laser-captured tumor samples will be subjected to low passage whole-genome, exome, and RNA sequencing (with targeted resequencing done in parallel), and reverse-phase protein and phosphorylation arrays, with circulating nucleic acids and circulating tumor cells being queried as well. “After that we hope to do a 100- or 150-patient trial when we have some idea of the best techniques,” he said.
Dr. Leyland-Jones predicted that ultimately most tumors will be found
- to have multiple drivers,
- with most patients receiving a combination of two, three, or perhaps four different targeted therapies.
Reduce to Practice
According to Randox, the evidence Investigator is a sophisticated semi-automated biochip system designed for research, clinical, forensic, and veterinary applications.
Once biomarkers that may have an impact on therapy are discovered, it is not always routine to get them into clinical practice. Leaving regulatory and financial, intellectual property and cultural issues aside, developing a diagnostic based on a biomarker often requires expertise or patience that its discoverer may not possess.
Andrew Gribben is a clinical assay and development scientist at Randox Laboratories, based in Northern Ireland, U.K. The company utilizes academic and industrial collaborators together with in-house discovery platforms to identify biomarkers that are
- augmented or diminished in a particular pathology
- relative to appropriate control populations.
Biomarkers can be developed to be run individually or
- combined into panels of immunoassays on its multiplex biochip array technology.
Specificity can also be gained—or lost—by the affinity of reagents in an assay. The diagnostic potential of Heart-type fatty acid binding protein (H-FABP) abundantly expressed in human myocardial cells was recognized by Jan Glatz of Maastricht University, The Netherlands, back in 1988. Levels rise quickly within 30 minutes after a myocardial infarction, peaking at 6–8 hours and return to normal within 24–30 hours. Yet at the time it was not known that H-FABP was a member of a multiprotein family, with which the polyclonal antibodies being used in development of an assay were cross-reacting, Gribben related.
Randox developed monoclonal antibodies specific to H-FABP, funded trials investigating its use alone, and multiplexed with cardiac biomarker assays, and, more than 30 years after the biomarker was identified, in 2011, released a validated assay for H-FABP as a biomarker for early detection of acute myocardial infarction.
Ultrasensitive Immunoassays for Biomarker Development
Research has shown that detection and monitoring of biomarker concentrations can provide
- insights into disease risk and progression.
Cytokines have become attractive biomarkers and candidates
- for targeted therapies for a number of autoimmune diseases, including rheumatoid arthritis (RA), Crohn’s disease, and psoriasis, among others.
However, due to the low-abundance of circulating cytokines, such as IL-17A, obtaining robust measurements in clinical samples has been difficult.
Singulex reports that its digital single-molecule counting technology provides
- increased precision and detection sensitivity over traditional ELISA techniques,
- helping to shed light on biomarker verification and validation programs.
The company’s Erenna® immunoassay system, which includes optimized immunoassays, offers LLoQ to femtogram levels per mL resolution—even in healthy populations, at an improvement of 1-3 fold over standard ELISAs or any conventional technology and with a dynamic range of up to 4-logs, according to a Singulex official, who adds that
- this sensitivity improvement helps minimize undetectable samples that
- could otherwise delay or derail clinical studies.
The official also explains that the Singulex solution includes an array of products and services that are being applied to a number of programs and have enabled the development of clinically relevant biomarkers, allowing translation from discovery to the clinic.
In a poster entitled “Advanced Single Molecule Detection: Accelerating Biomarker Development Utilizing Cytokines through Ultrasensitive Immunoassays,” a case study was presented of work performed by Jeff Greenberg of NYU to show how the use of the Erenna system can provide insights toward
- improving the clinical utility of biomarkers and
- accelerating the development of novel therapies for treating inflammatory diseases.
A panel of inflammatory biomarkers was examined in DMARD (disease modifying antirheumatic drugs)-naïve RA (rheumatoid arthritis) vs. knee OA (osteoarthritis) patient cohorts. Markers that exhibited significant differences in plasma concentrations between the two cohorts included
- CRP, IL-6R alpha, IL-6, IL-1 RA, VEGF, TNF-RII, and IL-17A, IL-17F, and IL-17A/F.
Among the three tested isoforms of IL-17,
- the magnitude of elevation for IL-17F in RA patients was the highest.
“Singulex provides high-resolution monitoring of baseline IL-17A concentrations that are present at low levels,” concluded the researchers. “The technology also enabled quantification of other IL-17 isoforms in RA patients, which have not been well characterized before.”
The Singulex Erenna System has also been applied to cardiovascular disease research, for which its
- cardiac troponin I (cTnI) digital assay can be used to measure circulating
- levels of cTnI undetectable by other commercial assays.
Recently presented data from Brigham and Women’s Hospital and the TIMI-22study showed that
- using the Singulex test to serially monitor cTnI helps
- stratify risk in post-acute coronary syndrome patients and
- can identify patients with elevated cTnI
- who have the most to gain from intensive vs. moderate-dose statin therapy,
according to the scientists involved in the research.
The study poster, “Prognostic Performance of Serial High Sensitivity Cardiac Troponin Determination in Stable Ischemic Heart Disease: Analysis From PROVE IT-TIMI 22,” was presented at the 2013 American College of Cardiology (ACC) Annual Scientific Session & Expo by R. O’Malley et al.
Biomarkers Changing Clinical Medicine
Better Diagnosis, Prognosis, and Drug Targeting Are among Potential Benefits
- John Morrow Jr., Ph.D.
Researchers at EMD Chemicals are developing biomarker immunoassays
- to monitor drug-induced toxicity including kidney damage.
The pace of biomarker development is accelerating as investigators report new studies on cancer, diabetes, Alzheimer disease, and other conditions in which the evaluation and isolation of workable markers is prominently featured.
Wei Zheng, Ph.D., leader of the R&D immunoassay group at EMD Chemicals, is overseeing a program to develop biomarker immunoassays to
- monitor drug-induced toxicity, including kidney damage.
“One of the principle reasons for drugs failing during development is because of organ toxicity,” says Dr. Zheng.
“proteins liberated into the serum and urine can serve as biomarkers of adverse response to drugs, as well as disease states.”
Through collaborative programs with Rules-Based Medicine (RBM), the EMD group has released panels for the profiling of human renal impairment and renal toxicity. These urinary biomarker based products fit the FDA and EMEA guidelines for assessment of drug-induced kidney damage in rats.
The group recently performed a screen for potential protein biomarkers in relation to
- kidney toxicity/damage on a set of urine and plasma samples
- from patients with documented renal damage.
Additionally, Dr. Zheng is directing efforts to move forward with the multiplexed analysis of
- organ and cellular toxicity.
Diseases thought to involve compromised oxidative phosphorylation include
- diabetes, Parkinson and Alzheimer diseases, cancer, and the aging process itself.
Good biomarkers allow Dr. Zheng to follow the mantra, “fail early, fail fast.” With robust, multiplexible biomarkers, EMD can detect bad drugs early and kill them before they move into costly large animal studies and clinical trials. “Recognizing the severe liability that toxicity presents, we can modify the structure of the candidate molecule and then rapidly reassess its performance.”
Scientists at Oncogene Science a division of Siemens Healthcare Diagnostics, are also focused on biomarkers. “We are working on a number of antibody-based tests for various cancers, including a test for the Ca-9 CAIX protein, also referred to ascarbonic anhydrase,” Walter Carney, Ph.D., head of the division, states.
CAIX is a transmembrane protein that is
- overexpressed in a number of cancers, and, like Herceptin and the Her-2 gene,
- can serve as an effective and specific marker for both diagnostic and therapeutic purposes.
- It is liberated into the circulation in proportion to the tumor burden.
Dr. Carney and his colleagues are evaluating patients after tumor removal for the presence of the Ca-9 CAIX protein. If
- the levels of the protein in serum increase over time,
- this suggests that not all the tumor cells were removed and the tumor has metastasized.
Dr. Carney and his team have developed both an immuno-histochemistry and an ELISA test that could be used as companion diagnostics in clinical trials of CAIX-targeted drugs.
The ELISA for the Ca-9 CAIX protein will be used in conjunction with Wilex’ Rencarex®, which is currently in a
- Phase III trial as an adjuvant therapy for non-metastatic clear cell renal cancer.
Additionally, Oncogene Science has in its portfolio an FDA-approved test for the Her-2 marker. Originally approved for Her-2/Neu-positive breast cancer, its indications have been expanded over time, and was approved
- for the treatment of gastric cancer last year.
It is normally present on breast cancer epithelia but
- overexpressed in some breast cancer tumors.
“Our products are designed to be used in conjunction with targeted therapies,” says Dr. Carney. “We are working with companies that are developing technology around proteins that are
- overexpressed in cancerous tissues and can be both diagnostic and therapeutic targets.”
The long-term goal of these studies is to develop individualized therapies, tailored for the patient. Since the therapies are expensive, accurate diagnostics are critical to avoid wasting resources on patients who clearly will not respond (or could be harmed) by the particular drug.
“At this time the rate of response to antibody-based therapies may be very poor, as
- they are often employed late in the course of the disease, and patients are in such a debilitated state
- that they lack the capacity to react positively to the treatment,” Dr. Carney explains.
Nanoscale Real-Time Proteomics
Stanford University School of Medicine researchers, working with Cell BioSciences, have developed a
- nanofluidic proteomic immunoassay that measures protein charge,
- similar to immunoblots, mass spectrometry, or flow cytometry.
- unlike these platforms, this approach can measure the amount of individual isoforms,
- specifically, phosphorylated molecules.
“We have developed a nanoscale device for protein measurement, which I believe could be useful for clinical analysis,” says Dean W. Felsher, M.D., Ph.D., associate professor at Stanford University School of Medicine.
Critical oncogenic transformations involving
- the activation of the signal-related kinases ERK-1 and ERK-2 can now be followed with ease.
“The fact that we measure nanoquantities with accuracy means that
- we can interrogate proteomic profiles in clinical patients,
by drawing tiny needle aspirates from tumors over the course of time,” he explains.
“This allows us to observe the evolution of tumor cells and
- their response to therapy
- from a baseline of the normal tissue as a standard of comparison.”
According to Dr. Felsher, 20 cells is a large enough sample to obtain a detailed description. The technology is easy to automate, which allows
- the inclusion of hundreds of assays.
Contrasting this technology platform with proteomic analysis using microarrays, Dr. Felsher notes that the latter is not yet workable for revealing reliable markers.
Dr. Felsher and his group published a description of this technology in Nature Medicine. “We demonstrated that we could take a set of human lymphomas and distinguish them from both normal tissue and other tumor types. We can
- quantify changes in total protein, protein activation, and relative abundance of specific phospho-isoforms
- from leukemia and lymphoma patients receiving targeted therapy.
Even with very small numbers of cells, we are able to show that the results are consistent, and
- our sample is a random profile of the tumor.”
Splice Variant Peptides
“Aberrations in alternative splicing may generate
- much of the variation we see in cancer cells,”
says Gilbert Omenn, Ph.D., director of the center for computational medicine and bioinformatics at the University of Michigan School of Medicine. Dr. Omenn and his colleague, Rajasree Menon, are
- using this variability as a key to new biomarker identification.
It is becoming evident that splice variants play a significant role in the properties of cancer cells, including
- initiation, progression, cell motility, invasiveness, and metastasis.
Alternative splicing occurs through multiple mechanisms
- when the exons or coding regions of the DNA transcribe mRNA,
- generating initiation sites and connecting exons in protein products.
Their translation into protein can result in numerous protein isoforms, and
- these isoforms may reflect a diseased or cancerous state.
Regulatory elements within the DNA are responsible for selecting different alternatives; thus
- the splice variants are tempting targets for exploitation as biomarkers.

Despite the many questions raised by these observations, splice variation in tumor material has not been widely studied. Cancer cells are known for their tremendous variability, which allows them to
- grow rapidly, metastasize, and develop resistance to anticancer drugs.
Dr. Omenn and his collaborators used
- mass spec data to interrogate a custom-built database of all potential mRNA sequences
- to find alternative splice variants.
When they compared normal and malignant mammary gland tissue from a mouse model of Her2/Neu human breast cancers, they identified a vast number (608) of splice variant proteins, of which
- peptides from 216 were found only in the tumor sample.
“These novel and known alternative splice isoforms
- are detectable both in tumor specimens and in plasma and
- represent potential biomarker candidates,” Dr. Omenn adds.
Dr. Omenn’s observations and those of his colleague Lewis Cantley, Ph.D., have also
- shed light on the origins of the classic Warburg effect,
- the shift to anaerobic glycolysis in tumor cells.
The novel splice variant M2, of muscle pyruvate kinase,
- is observed in embryonic and tumor tissue.
It is associated with this shift, the result of
- the expression of a peptide splice variant sequence.
It is remarkable how many different areas of the life sciences are tied into the phenomenon of splice variation. The changes in the genetic material can be much greater than point mutations, which have been traditionally considered to be the prime source of genetic variability.
“We now have powerful methods available to uncover a whole new category of variation,” Dr. Omenn says. “High-throughput RNA sequencing and proteomicswill be complementary in discovery studies of splice variants.”
Splice variation may play an important role in rapid evolutionary changes, of the sort discussed by Susumu Ohno and Stephen J. Gould decades ago. They, and other evolutionary biologists, argued that
- gene duplication, combined with rapid variability, could fuel major evolutionary jumps.
At the time, the molecular mechanisms of variation were poorly understood, but today
- the tools are available to rigorously evaluate the role of
- splice variation and other contributors to evolutionary change.
“Biomarkers derived from studies of splice variants, could, in the future, be exploited
- both for diagnosis and prognosis and
- for drug targeting of biological networks,
- in situations such as the Her-2/Neu breast cancers,” Dr. Omenn says.
Aminopeptidase Activities
“By correlating the proteolytic patterns with disease groups and controls, we have shown that
- exopeptidase activities contribute to the generation of not only cancer-specific
- but also cancer type specific serum peptides.
according to Paul Tempst, Ph.D., professor and director of the Protein Center at the Memorial Sloan-Kettering Cancer Center.
So there is a direct link between peptide marker profiles of disease and differential protease activity.” For this reason Dr. Tempst argues that “the patterns we describe may have value as surrogate markers for detection and classification of cancer.”
To investigate this avenue, Dr. Tempst and his colleagues have followed
- the relationship between exopeptidase activities and metastatic disease.
“We monitored controlled, de novo peptide breakdown in large numbers of biological samples using mass spectrometry, with relative quantitation of the metabolites,” Dr. Tempst explains. This entailed the use of magnetic, reverse-phase beads for analyte capture and a MALDI-TOF MS read-out.
“In biomarker discovery programs, functional proteomics is usually not pursued,” says Dr. Tempst. “For putative biomarkers, one may observe no difference in quantitative levels of proteins, while at the same time, there may be substantial differences in enzymatic activity.”
In a preliminary prostate cancer study, the team found a significant difference
- in activity levels of exopeptidases in serum from patients with metastatic prostate cancer
- as compared to primary tumor-bearing individuals and normal healthy controls.
However, there were no differences in amounts of the target protein, and this potential biomarker would have been missed if quantitative levels of protein had been the only criterion of selection.
It is frequently stated that “practical fusion energy is 30 years in the future and always will be.” The same might be said of functional, practical biomarkers that can pass muster with the FDA. But splice variation represents a new handle on this vexing problem. It appears that we are seeing the emergence of a new approach that may finally yield definitive diagnostic tests, detectable in serum and urine samples.
Part 7
Epigenetics and Drug Metabolism
DNA Methylation Rules: Studying Epigenetics with New Tools
The tools to unravel the epigenetic control mechanisms that influence how cells control access of transcriptional proteins to DNA are just beginning to emerge.
Patricia Fitzpatrick Dimond, Ph.D.
{kind=link}
New tools may help move the field of epigenetic analysis forward and potentially unveil novel biomarkers for cellular development, differentiation, and disease.
DNA sequencing has had the power of technology behind it as novel platforms to produce more sequencing faster and at lower cost have been introduced. But the tools to unravel the epigenetic control mechanisms that influence how cells control access of transcriptional proteins to DNA are just beginning to emerge.
Among these mechanisms, DNA methylation, or the enzymatically mediated addition of a methyl group to cytosine or adenine dinucleotides,
- serves as an inherited epigenetic modification that
- stably modifies gene expression in dividing cells.
The unique methylomes are largely maintained in differentiated cell types, making them critical to understanding the differentiation potential of the cell.
In the DNA methylation process, cytosine residues in the genome are enzymatically modified to 5-methylcytosine,
- which participates in transcriptional repression of genes during development and disease progression.
5-methylcytosine can be further enzymatically modified to 5-hydroxymethylcytosine by the TET family of methylcytosine dioxygenases. DNA methylation affects gene transcription by physically
- interfering with the binding of proteins involved in gene transcription.
Methylated DNA may be bound by methyl-CpG-binding domain proteins (MBDs) that can
- then recruit additional proteins. Some of these include histone deacetylases and other chromatin remodeling proteins that modify histones, thereby
- forming compact, inactive chromatin, or heterochromatin.
While DNA methylation doesn’t change the genetic code,
- it influences chromosomal stability and gene expression.
Epigenetics and Cancer Biomarkers

And because of the increasing recognition that DNA methylation changes are involved in human cancers, scientists have suggested that these epigenetic markers may provide biological markers for cancer cells, and eventually point toward new diagnostic and therapeutic targets. Cancer cell genomes display genome-wide abnormalities in DNA methylation patterns,
- some of which are oncogenic and contribute to genome instability.
In particular, de novo methylation of tumor suppressor gene promoters
- occurs frequently in cancers, thereby silencing them and promoting transformation.
Cytosine hydroxymethylation (5-hydroxymethylcytosine, or 5hmC), the aforementioned DNA modification resulting from the enzymatic conversion of 5mC into 5-hydroxymethylcytosine by the TET family of oxygenases, has been identified
- as another key epigenetic modification marking genes important for
- pluripotency in embryonic stem cells (ES), as well as in cancer cells.
The base 5-hydroxymethylcytosine was recently identified as an oxidation product of 5-methylcytosine in mammalian DNA. In 2011, using sensitive and quantitative methods to assess levels of 5-hydroxymethyl-2′-deoxycytidine (5hmdC) and 5-methyl-2′-deoxycytidine (5mdC) in genomic DNA, scientists at the Department of Cancer Biology, Beckman Research Institute of the City of Hope, Duarte, California investigated
- whether levels of 5hmC can distinguish normal tissue from tumor tissue.
They showed that in squamous cell lung cancers, levels of 5hmdC showed
- up to five-fold reduction compared with normal lung tissue.
In brain tumors,5hmdC showed an even more drastic reduction
- with levels up to more than 30-fold lower than in normal brain,
- but 5hmdC levels were independent of mutations in isocitrate dehydrogenase-1, the enzyme that converts 5hmC to 5hmdC.
Immunohistochemical analysis indicated that 5hmC is “remarkably depleted” in many types of human cancer.
- there was an inverse relationship between 5hmC levels and cell proliferation with lack of 5hmC in proliferating cells.
Their data suggest that 5hmdC is strongly depleted in human malignant tumors,
- a finding that adds another layer of complexity to the aberrant epigenome found in cancer tissue.
In addition, a lack of 5hmC may become a useful biomarker for cancer diagnosis.
Enzymatic Mapping
But according to New England Biolabs’ Sriharsa Pradhan, Ph.D., methods for distinguishing 5mC from 5hmC and analyzing and quantitating the cell’s entire “methylome” and “hydroxymethylome” remain less than optimal.
The protocol for bisulphite conversion to detect methylation remains the “gold standard” for DNA methylation analysis. This method is generally followed by PCR analysis for single nucleotide resolution to determine methylation across the DNA molecule. According to Dr. Pradhan, “.. bisulphite conversion does not distinguish 5mC and 5hmC,”
Recently we found an enzyme, a unique DNA modification-dependent restriction endonuclease, AbaSI, which can
- decode the hydryoxmethylome of the mammalian genome.
You easily can find out where the hydroxymethyl regions are.”
AbaSI, recognizes 5-glucosylatedmethylcytosine (5gmC) with high specificity when compared to 5mC and 5hmC, and
- cleaves at narrow range of distances away from the recognized modified cytosine.
By mapping the cleaved ends, the exact 5hmC location can, the investigators reported, be determined.
Dr. Pradhan and his colleagues at NEB; the Department of Biochemistry, Emory University School of Medicine, Atlanta; and the New England Biolabs Shanghai R&D Center described use of this technique in a paper published in Cell Reports this month, in which they described high-resolution enzymatic mapping of genomic hydroxymethylcytosine in mouse ES cells.
In the current report, the authors used the enzyme technology for the genome-wide high-resolution hydroxymethylome, describing simple library construction even with a low amount of input DNA (50 ng) and the ability to readily detect 5hmC sites with low occupancy.
As a result of their studies, they propose that
factors affecting the local 5mC accessibility to TET enzymes play important roles in the 5hmC deposition
- including include chromatin compaction, nucleosome positioning, or TF binding.
- the regularly oscillating 5hmC profile around the CTCF-binding sites, suggests 5hmC ‘‘writers’’ may be sensitive to the nucleosomal environment.
- some transiently stable 5hmCs may indicate a poised epigenetic state or demethylation intermediate, whereas others may suggest a locally accessible chromosomal environment for the TET enzymatic apparatus.
“We were able to do complete mapping in mouse embryonic cells and are pleased about what this enzyme can do and how it works,” Dr. Pradhan said.
And the availability of novel tools that make analysis of the methylome and hypomethylome more accessible will move the field of epigenetic analysis forward and potentially novel biomarkers for cellular development, differentiation, and disease.
Patricia Fitzpatrick Dimond, Ph.D. (pdimond@genengnews.com), is technical editor at Genetic Engineering & Biotechnology News.
Epigenetic Regulation of ADME-Related Genes: Focus on Drug Metabolism and Transport
Published: Sep 23, 2013
Epigenetic regulation of gene expression refers to heritable factors that are functionally relevant genomic modifications but that do not involve changes in DNA sequence.
Examples of such modifications include
- DNA methylation, histone modifications, noncoding RNAs, and chromatin architecture.
Epigenetic modifications are crucial for
packaging and interpreting the genome, and they have fundamental functions in regulating gene expression and activity under the influence of physiologic and environmental factors.
In this issue of Drug Metabolism and Disposition, a series of articles is presented to demonstrate the role of epigenetic factors in regulating
- the expression of genes involved in drug absorption, distribution, metabolism, and excretion in organ development, tissue-specific gene expression, sexual dimorphism, and in the adaptive response to xenobiotic exposure, both therapeutic and toxic.
The articles also demonstrate that, in addition to genetic polymorphisms, epigenetics may also contribute to wide inter-individual variations in drug metabolism and transport. Identification of functionally relevant epigenetic biomarkers in human specimens has the potential to improve prediction of drug responses based on patient’s epigenetic profiles.
http://www.technologynetworks.com/Metabolomics/news.aspx?ID=157804
This study is published online in Drug Metabolism and Disposition
Part 8
Pictorial Maps
Prediction of intracellular metabolic states from extracellular metabolomic data
MK Aurich, G Paglia, Ottar Rolfsson, S Hrafnsdottir, M Magnusdottir, MM Stefaniak, BØ Palsson, RMT Fleming &
Ines Thiele
Metabolomics Aug 14, 2014;
http://dx.doi.org:/10.1007/s11306-014-0721-3
http://link.springer.com/article/10.1007/s11306-014-0721-3/fulltext.html#Sec1
{kind=link}
Metabolic models can provide a mechanistic framework
- to analyze information-rich omics data sets, and are
- increasingly being used to investigate metabolic alternations in human diseases.
An expression of the altered metabolic pathway utilization is the selection of metabolites consumed and released by cells. However, methods for the
- inference of intracellular metabolic states from extracellular measurements in the context of metabolic models remain underdeveloped compared to methods for other omics data.
Herein, we describe a workflow for such an integrative analysis
- emphasizing on extracellular metabolomics data.
We demonstrate,
- using the lymphoblastic leukemia cell lines Molt-4 and CCRF-CEM,
how our methods can reveal differences in cell metabolism. Our models explain metabolite uptake and secretion by predicting
- a more glycolytic phenotype for the CCRF-CEM model and
- a more oxidative phenotype for the Molt-4 model,
- which was supported by our experimental data.
Gene expression analysis revealed altered expression of gene products at
- key regulatory steps in those central metabolic pathways, and
literature query emphasized the role of these genes in cancer metabolism.
Moreover, in silico gene knock-outs identified unique
- control points for each cell line model, e.g., phosphoglycerate dehydrogenase for the Molt-4 model.
Thus, our workflow is well suited to the characterization of cellular metabolic traits based on
- -extracellular metabolomic data, and it allows the integration of multiple omics data sets
- into a cohesive picture based on a defined model context.
Keywords Constraint-based modeling _ Metabolomics _ Multi-omics _ Metabolic network _ Transcriptomics
1 Introduction
Modern high-throughput techniques have increased the pace of biological data generation. Also referred to as the ‘‘omics avalanche’’, this wealth of data provides great opportunities for metabolic discovery. Omics data sets
- contain a snapshot of almost the entire repertoire of mRNA, protein, or metabolites at a given time point or
under a particular set of experimental conditions. Because of the high complexity of the data sets,
- computational modeling is essential for their integrative analysis.
Currently, such data analysis is a bottleneck in the research process and methods are needed to facilitate the use of these data sets, e.g., through meta-analysis of data available in public databases [e.g., the human protein atlas (Uhlen et al. 2010) or the gene expression omnibus (Barrett et al. 2011)], and to increase the accessibility of valuable information for the biomedical research community.
Constraint-based modeling and analysis (COBRA) is
- a computational approach that has been successfully used to
- investigate and engineer microbial metabolism through the prediction of steady-states (Durot et al.2009).
The basis of COBRA is network reconstruction: networks are assembled in a bottom-up fashion based on
- genomic data and extensive
- organism-specific information from the literature.
Metabolic reconstructions capture information on the
- known biochemical transformations taking place in a target organism
- to generate a biochemical, genetic and genomic knowledge base (Reed et al. 2006).
Once assembled, a
- metabolic reconstruction can be converted into a mathematical model (Thiele and Palsson 2010), and
- model properties can be interrogated using a great variety of methods (Schellenberger et al. 2011).
The ability of COBRA models
- to represent genotype–phenotype and environment–phenotype relationships arises
- through the imposition of constraints, which
- limit the system to a subset of possible network states (Lewis et al. 2012).
Currently, COBRA models exist for more than 100 organisms, including humans (Duarte et al. 2007; Thiele et al. 2013).
Since the first human metabolic reconstruction was described [Recon 1 (Duarte et al. 2007)],
- biomedical applications of COBRA have increased (Bordbar and Palsson 2012).
One way to contextualize networks is to
- define their system boundaries according to the metabolic states of the system, e.g., disease or dietary regimes.
The consequences of the applied constraints can
- then be assessed for the entire network (Sahoo and Thiele 2013).
Additionally, omics data sets have frequently been used
- to generate cell-type or condition-specific metabolic models.
Models exist for specific cell types, such as
- enterocytes (Sahoo and Thiele2013),
- macrophages (Bordbar et al. 2010),
- adipocytes (Mardinoglu et al. 2013),
- even multi-cell assemblies that represent the interactions of brain cells (Lewis et al. 2010).
All of these cell type specific models, except the enterocyte reconstruction
- were generated based on omics data sets.
Cell-type-specific models have been used to study
- diverse human disease conditions.
For example, an adipocyte model was generated using
- transcriptomic, proteomic, and metabolomics data.
This model was subsequently used to investigate metabolic alternations in adipocytes
- that would allow for the stratification of obese patients (Mardinoglu et al. 2013).
The biomedical applications of COBRA have been
- cancer metabolism (Jerby and Ruppin, 2012).
- predicting drug targets (Folger et al. 2011; Jerby et al. 2012).
A cancer model was generated using
- multiple gene expression data sets and subsequently used
- to predict synthetic lethal gene pairs as potential drug targets
- selective for the cancer model, but non-toxic to the global model (Recon 1),
a consequence of the reduced redundancy in the cancer specific model (Folger et al. 2011).
In a follow up study, lethal synergy between FH and enzymes of the heme metabolic pathway
- were experimentally validated and resolved the mechanism by which FH deficient cells,
e.g., in renal-cell cancer cells survive a non-functional TCA cycle (Frezza et al. 2011).
Contextualized models, which contain only the subset of reactions active in a particular tissue (or cell-) type,
- can be generated in different ways (Becker and Palsson, 2008; Jerby et al. 2010).
However, the existing algorithms mainly consider
- gene expression and proteomic data
- to define the reaction sets that comprise the contextualized metabolic models.
These subset of reactions are usually defined
- based on the expression or absence of expression of the genes or proteins (present and absent calls),
- or inferred from expression values or differential gene expression.
Comprehensive reviews of the methods are available (Blazier and Papin, 2012; Hyduke et al. 2013). Only the compilation of a large set of omics data sets
- can result in a tissue (or cell-type) specific metabolic model, whereas
the representation of one particular experimental condition is achieved
- through the integration of omics data set generated from one experiment only (condition-specific cell line model).
Recently, metabolomic data sets have become more comprehensive and
- using these data sets allow direct determination of the metabolic network components (the metabolites).
Additionally, metabolomics has proven to be stable, relatively inexpensive, and highly reproducible (Antonucci et al. 2012). These factors make metabolomic data sets particularly valuable for
- interrogation of metabolic phenotypes.
Thus, the integration of these data sets is now an active field of research (Li et al. 2013; Mo et al. 2009; Paglia et al. 2012b; Schmidt et al. 2013).
Generally, metabolomic data can be incorporated into metabolic networks as
- qualitative, quantitative, and thermodynamic constraints (Fleming et al. 2009; Mo et al. 2009).
Mo et al. used metabolites detected in the
- spent medium of yeast cells to determine intracellular flux states through a sampling analysis (Mo et al. 2009),
- which allowed unbiased interrogation of the possible network states (Schellenberger and Palsson 2009) and
- prediction of internal pathway use.

Such analyses have also been used to reveal the effects of
- enzymopathies on red blood cells (Price et al. 2004),
- to study effects of diet on diabetes (Thiele et al. 2005) and
- to define macrophage metabolic states (Bordbar et al. 2010).
This type of analysis is available as a function in the COBRA toolbox (Schellenberger et al. 2011).
In this study, we established a workflow
- for the generation and analysis of condition-specific metabolic cell line models
- that can facilitate the interpretation of metabolomic data.
Our modeling yields meaningful predictions regarding
- metabolic differences between two lymphoblastic leukemia cell lines (Fig. 1A).
Fig. 1

A Combined experimental and computational pipeline to study human metabolism.
- Experimental work and omics data analysis steps precede computational modeling.
- Model predictions are validated based on targeted experimental data.
- Metabolomic and transcriptomic data are used for model refinement and submodel extraction.
- Functional analysis methods are used to characterize the metabolism of the cell-line models and compare it to additional experimental data.
- The validated models are subsequently used for the prediction of drug targets.
B Uptake and secretion pattern of model metabolites. All metabolite uptakes and secretions that were mapped during model generation are shown.
- Metabolite uptakes are depicted on the left, and
- secreted metabolites are shown on the right.
- A number of metabolite exchanges mapped to the model wereunique to one cell line.
- Differences between cell lines were used to set quantitative constraints for the sampling analysis.
C Statistics about the cell line-specific network generation.
D Quantitative constraints.
For the sampling analysis, an additional set of constraints was imposed on the cell line specific models,
- emphasizing the differences in metabolite uptake and secretion between cell lines.
Higher uptake of a metabolite was allowed
- in the model of the cell line that consumed more of the metabolite in vitro, whereas
- the supply was restricted for the model with lower in vitro uptake.
This was done by establishing the same ratio between the models bounds as detected in vitro.
X denotes the factor (slope ratio) that distinguishes the bounds, and
- which was individual for each metabolite.
(a) The uptake of a metabolite could be x times higher in CCRF-CEM cells,
(b) the metabolite uptake could be x times higher in Molt-4,
(c) metabolite secretion could be x times higher in CCRF-CEM, or
(d) metabolite secretion could be x times higher in Molt-4 cells.LOD limit of detection.
The consequence of the adjustment was, in case of uptake, that one model was constrained to a lower metabolite uptake (A, B), and the difference depended on the ratio detected in vitro. In case of secretion, one model
- had to secrete more of the metabolite, and again
- the difference depended on the experimental difference detected between the cell lines
2 Results
We set up a pipeline that could be used to infer intracellular metabolic states
- from semi-quantitative data regarding metabolites exchanged between cells and their environment.
Our pipeline combined the following four steps:
- data acquisition,
- data analysis,
- metabolic modeling and
- experimental validation of the model predictions (Fig. 1A).
We demonstrated the pipeline and the predictive potential to predict metabolic alternations in diseases such as cancer based on
^two lymphoblastic leukemia cell lines.
The resulting Molt-4 and CCRF-CEM condition-specific cell line models could explain
^ metabolite uptake and secretion
^ by predicting the distinct utilization of central metabolic pathways by the two cell lines.
^ the CCRF-CEM model resembled more a glycolytic, commonly referred to as ‘Warburg’ phenotype,
^ our model predicted a more respiratory phenotype for the Molt-4 model.
We found these predictions to be in agreement with measured gene expression differences
- at key regulatory steps in the central metabolic pathways, and they were also
- consistent with additional experimental data regarding the energy and redox states of the cells.
After a brief discussion of the data generation and analysis steps, the results derived from model generation and analysis will be described in detail.
2.1 Pipeline for generation of condition-specific metabolic cell line models

2.1.1 Generation of experimental data
We monitored the growth and viability of lymphoblastic leukemia cell lines in serum-free medium (File S2, Fig. S1). Multiple omics data sets were derived from these cells.Extracellular metabolomics (exo-metabolomic) data,
^ comprising measurements of the metabolites in the spent medium of the cell cultures (Paglia et al. 2012a),
^ were collected along with transcriptomic data, and these data sets were used to construct the models.
2.1.4 Condition-specific models for CCRF-CEM and Molt-4 cells
To determine whether we had obtained two distinct models, we evaluated the reactions, metabolites, and genes of the two models. Both the Molt-4 and CCRF-CEM models contained approximately half of the reactions and metabolites present in the global model (Fig. 1C). They were very similar to each other in terms of their reactions, metabolites, and genes (File S1, Table S5A–C).
(1) The Molt-4 model contained seven reactions that were not present in the CCRF-CEM model (Co-A biosynthesis pathway and exchange reactions).
(2) The CCRF-CEM contained 31 unique reactions (arginine and proline metabolism, vitamin B6 metabolism, fatty acid activation, transport, and exchange reactions).
(3) There were 2 and 15 unique metabolites in the Molt-4 and CCRF-CEM models, respectively (File S1, Table S5B).
(4) Approximately three quarters of the global model genes remained in the condition-specific cell line models (Fig. 1C).
(5) The Molt-4 model contained 15 unique genes, and the CCRF-CEM model had 4 unique genes (File S1, Table S5C).
(6) Both models lacked NADH dehydrogenase (complex I of the electron transport chain—ETC), which was determined by the absence of expression of a mandatory subunit (NDUFB3, Entrez gene ID 4709).
Rather, the ETC was fueled by FADH2 originating from succinate dehydrogenase and from fatty acid oxidation, which through flavoprotein electron transfer

- could contribute to the same ubiquinone pool as complex I and complex II (succinate dehydrogenase).
Despite their different in vitro growth rates (which differed by 11 %, see File S2, Fig. S1) and
^^^ differences in exo-metabolomic data (Fig. 1B) and transcriptomic data,
^^^ the internal networks were largely conserved in the two condition-specific cell line models.
2.1.5 Condition-specific cell line models predict distinct metabolic strategies
Despite the overall similarity of the metabolic models, differences in their cellular uptake and secretion patterns suggested distinct metabolic states in the two cell lines (Fig. 1B and see “Materials and methods” section for more detail). To interrogate the metabolic differences, we sampled the solution space of each model using an Artificial Centering Hit-and-Run (ACHR) sampler (Thiele et al. 2005). For this analysis, additional constraints were applied, emphasizing the quantitative differences in commonly uptaken and secreted metabolites. The maximum possible uptake and maximum possible secretion flux rates were reduced
^^^ according to the measured relative differences between the cell lines (Fig. 1D, see “Materials and methods” section).
We plotted the number of sample points containing a particular flux rate for each reaction. The resulting binned histograms can be understood as representing the probability that a particular reaction can have a certain flux value.
A comparison of the sample points obtained for the Molt-4 and CCRF-CEM models revealed
- a considerable shift in the distributions, suggesting a higher utilization of glycolysis by the CCRF-CEM model
(File S2, Fig. S2).
This result was further supported by differences in medians calculated from sampling points (File S1, Table S6).
The shift persisted throughout all reactions of the pathway and was induced by thehigher glucose uptake (34 %) from the extracellular medium in CCRF-CEM cells.
The sampling median for glucose uptake was 34 % higher in the CCRF-CEM model than in Molt-4 model (File S2, Fig. S2).
The usage of the TCA cycle was also distinct in the two condition-specific cell-line models (Fig. 2). Interestingly,
the models used succinate dehydrogenase differently (Figs. 2, 3).

The Molt-4 model utilized an associated reaction to generate FADH2, whereas
- in the CCRF-CEM model, the histogram was shifted in the opposite direction,
- toward the generation of succinate.
Additionally, there was a higher efflux of citrate toward amino acid and lipid metabolism in the CCRF-CEM model (Fig. 2). There was higher flux through anaplerotic and cataplerotic reactions in the CCRF-CEM model than in the Molt-4 model (Fig. 2); these reactions include
(1) the efflux of citrate through ATP-citrate lyase,
(2) uptake of glutamine,
(3) generation of glutamate from glutamine,
(4) transamination of pyruvate and glutamate to alanine and to 2-oxoglutarate,
(5) secretion of nitrogen, and
(6) secretion of alanine.

The Molt-4 model showed higher utilization of oxidative phosphorylation(Fig. 3), again supported by
elevated median flux through ATP synthase (36 %) and other enzymes, which contributed to higher oxidative metabolism. The sampling analysis therefore revealed different usage of central metabolic pathways by the condition-specific models.
Fig. 2

Differences in the use of the TCA cycle by the CCRF-CEM model (red) and the Molt-4 model (blue).
The table provides the median values of the sampling results. Negative values in histograms and in the table describe reversible reactions with flux in the reverse direction. There are multiple reversible reactions for the transformation of isocitrate and α-ketoglutarate, malate and fumarate, and succinyl-CoA and succinate. These reactions are unbounded, and therefore histograms are not shown. The details of participating cofactors have been removed.
Figure 3.

Atp ATP, cit citrate, adp ADP, pi phosphate, oaa oxaloacetate, accoa acetyl-CoA, coa coenzyme-A, icit isocitrate, αkg α-ketoglutarate, succ-coa succinyl-CoA, succ succinate, fumfumarate, mal malate, oxa oxaloacetate,
pyr pyruvate, lac lactate, ala alanine, gln glutamine, ETC electron transport chain









I actually consider this amazing blog , âSAME SCIENTIFIC IMPACT: Scientific Publishing –
Open Journals vs. Subscription-based « Pharmaceutical Intelligenceâ, very compelling plus the blog post ended up being a good read.
Many thanks,Annette
I actually consider this amazing blog , âSAME SCIENTIFIC IMPACT: Scientific Publishing –
Open Journals vs. Subscription-based « Pharmaceutical Intelligenceâ, very compelling plus the blog post ended up being a good read.
Many thanks,Annette
I actually consider this amazing blog , âSAME SCIENTIFIC IMPACT: Scientific Publishing –
Open Journals vs. Subscription-based « Pharmaceutical Intelligenceâ, very compelling plus the blog post ended up being a good read.
Many thanks,Annette
I actually consider this amazing blog , âSAME SCIENTIFIC IMPACT: Scientific Publishing –
Open Journals vs. Subscription-based « Pharmaceutical Intelligenceâ, very compelling plus the blog post ended up being a good read.
Many thanks,Annette