Funding, Deals & Partnerships: BIOLOGICS & MEDICAL DEVICES; BioMed e-Series; Medicine and Life Sciences Scientific Journal – http://PharmaceuticalIntelligence.com
Medical Startups – Artificial Intelligence (AI) Startups in Healthcare
Reporters: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN and Shraga Rottem, MD, DSc,
The motivation for this post is two fold:
First, we are presenting an application of AI, NLP, DL to our own medical text in the Genomics space. Here we present the first section of Part 1 in the following book. Part 1 has six subsections that yielded 12 plots. The entire Book is represented by 38 x 2 = 76 plots.
Second, we bring to the attention of the e-Reader the list of 276 Medical Startups – Artificial Intelligence (AI) Startups in Healthcare as a hot universe of R&D activity in Human Health.
Third, to highlight one academic center with an AI focus
Dear friends of the ETH AI Center,
We would like to provide you with some exciting updates from the ETH AI Center and its growing community.
As the Covid-19 restrictions in Switzerland have recently been lifted, we would like to hear from you what kind of events you would like to see in 2022! Participate in the survey to suggest event formats and topics that you would enjoy being a part of. We are already excited to learn what we can achieve together this year.
We already have many interesting events coming up, we look forward to seeing you at our main and community events!
LPBI Group is applying AI for Medical Text Analysis with Machine Learning and Natural Language Processing: Statistical and Deep Learning
Our Book
Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS & BioInformatics, Simulations and the Genome Ontology
Medical Text Analysis of this Books shows the following results obtained by Madison Davis by applying Wolfram NLP for Biological Languages on our own Text. See below an Example:
@MIT Artificial intelligence system rapidly predicts how two proteins will attach: The model called Equidock, focuses on rigid body docking — which occurs when two proteins attach by rotating or translating in 3D space, but their shapes don’t squeeze or bend
Reporter: Aviva Lev-Ari, PhD, RN
This paper introduces a novel SE(3) equivariant graph matching network, along with a keypoint discovery and alignment approach, for the problem of protein-protein docking, with a novel loss based on optimal transport. The overall consensus is that this is an impactful solution to an important problem, whereby competitive results are achieved without the need for templates, refinement, and are achieved with substantially faster run times.
Keywords:protein complexes, protein structure, rigid body docking, SE(3) equivariance, graph neural networks
Abstract: Protein complex formation is a central problem in biology, being involved in most of the cell’s processes, and essential for applications such as drug design or protein engineering. We tackle rigid body protein-protein docking, i.e., computationally predicting the 3D structure of a protein-protein complex from the individual unbound structures, assuming no three-dimensional flexibility during binding. We design a novel pairwise-independent SE(3)-equivariant graph matching network to predict the rotation and translation to place one of the proteins at the right location and the right orientation relative to the second protein. We mathematically guarantee that the predicted complex is always identical regardless of the initial placements of the two structures, avoiding expensive data augmentation. Our model approximates the binding pocket and predicts the docking pose using keypoint matching and alignment through optimal transport and a differentiable Kabsch algorithm. Empirically, we achieve significant running time improvements over existing protein docking software and predict qualitatively plausible protein complex structures despite not using heavy sampling, structure refinement, or templates.
One-sentence Summary: We perform rigid protein docking using a novel independent SE(3)-equivariant message passing mechanism that guarantees the same resulting protein complex independent of the initial placement of the two 3D structures.
MIT researchers created a machine-learning model that can directly predict the complex that will form when two proteins bind together. Their technique is between 80 and 500 times faster than state-of-the-art software methods, and often predicts protein structures that are closer to actual structures that have been observed experimentally.
This technique could help scientists better understand some biological processes that involve protein interactions, like DNA replication and repair; it could also speed up the process of developing new medicines.
“Deep learning is very good at capturing interactions between different proteins that are otherwise difficult for chemists or biologists to write experimentally. Some of these interactions are very complicated, and people haven’t found good ways to express them. This deep-learning model can learn these types of interactions from data,” says Octavian-Eugen Ganea, a postdoc in the MIT Computer Science and Artificial Intelligence Laboratory (CSAIL) and co-lead author of the paper.
Ganea’s co-lead author is Xinyuan Huang, a graduate student at ETH Zurich. MIT co-authors include Regina Barzilay, the School of Engineering Distinguished Professor for AI and Health in CSAIL, and Tommi Jaakkola, the Thomas Siebel Professor of Electrical Engineering in CSAIL and a member of the Institute for Data, Systems, and Society. The research will be presented at the International Conference on Learning Representations.
Significance of the Scientific Development by the @MIT Team
EquiDock wide applicability:
Our method can be integrated end-to-end to boost the quality of other models (see above discussion on runtime importance). Examples are predicting functions of protein complexes [3] or their binding affinity [5], de novo generation of proteins binding to specific targets (e.g., antibodies [6]), modeling back-bone and side-chain flexibility [4], or devising methods for non-binary multimers. See the updated discussion in the “Conclusion” section of our paper.
Advantages over previous methods:
Our method does not rely on templates or heavy candidate sampling [7], aiming at the ambitious goal of predicting the complex pose directly. This should be interpreted in terms of generalization (to unseen structures) and scalability capabilities of docking models, as well as their applicability to various other tasks (discussed above).
Our method obtains a competitive quality without explicitly using previous geometric (e.g., 3D Zernike descriptors [8]) or chemical (e.g., hydrophilic information) features [3]. Future EquiDock extensions would find creative ways to leverage these different signals and, thus, obtain more improvements.
Novelty of theory:
Our work is the first to formalize the notion of pairwise independent SE(3)-equivariance. Previous work (e.g., [9,10]) has incorporated only single object Euclidean-equivariances into deep learning models. For tasks such as docking and binding of biological objects, it is crucial that models understand the concept of multi-independent Euclidean equivariances.
All propositions in Section 3 are our novel theoretical contributions.
We have rewritten the Contribution and Related Work sections to clarify this aspect.
Footnote [a]: We have fixed an important bug in the cross-attention code. We have done a more extensive hyperparameter search and understood that layer normalization is crucial in layers used in Eqs. 5 and 9, but not on the h embeddings as it was originally shown in Eq. 10. We have seen benefits from training our models with a longer patience in the early stopping criteria (30 epochs for DIPS and 150 epochs for DB5). Increasing the learning rate to 2e-4 is important to speed-up training. Using an intersection loss weight of 10 leads to improved results compared to the default of 1.
Bibliography:
[1] Protein-ligand blind docking using QuickVina-W with inter-process spatio-temporal integration, Hassan et al., 2017
[2] GNINA 1.0: molecular docking with deep learning, McNutt et al., 2021
[3] Protein-protein and domain-domain interactions, Kangueane and Nilofer, 2018
[4] Side-chain Packing Using SE(3)-Transformer, Jindal et al., 2022
[5] Contacts-based prediction of binding affinity in protein–protein complexes, Vangone et al., 2015
[6] Iterative refinement graph neural network for antibody sequence-structure co-design, Jin et al., 2021
[7] Hierarchical, rotation-equivariant neural networks to select structural models of protein complexes, Eismann et al, 2020
[8] Protein-protein docking using region-based 3D Zernike descriptors, Venkatraman et al., 2009
[9] SE(3)-transformers: 3D roto-translation equivariant attention networks, Fuchs et al, 2020
[10] E(n) equivariant graph neural networks, Satorras et al., 2021
[11] Fast end-to-end learning on protein surfaces, Sverrisson et al., 2020
Big pharma companies are snapping up collaborations with firms using AI to speed up drug discovery, with one of the latest being Sanofi’s pact with Exscientia.
Tech giants are placing big bets on digital health analysis firms, such as Oracle’s €25.42B ($28.3B) takeover of Cerner in the US.
There’s also a steady flow of financing going to startups taking new directions with AI and bioinformatics, with the latest example being a €20M Series A round by SeqOne Genomics in France.
“IBM Watson uses a philosophy that is diametrically opposed to SeqOne’s,” said Jean-Marc Holder, CSO of SeqOne. “[IBM Watson seems] to rely on analysis of large amounts of relatively unstructured data and bet on the volume of data delivering the right result. By opposition, SeqOne strongly believes that data must be curated and structured in order to deliver good results in genomics.”
Francisco Partners is picking up a range of databases and analytics tools – including
Health Insights,
MarketScan,
Clinical Development,
Social Programme Management,
Micromedex and
other imaging and radiology tools, for an undisclosed sum estimated to be in the region of $1 billion.
IBM said the sell-off is tagged as “a clear next step” as it focuses on its platform-based hybrid cloud and artificial intelligence strategy, but it’s no secret that Watson Health has failed to live up to its early promise.
The sale also marks a retreat from healthcare for the tech giant, which is remarkable given that it once said it viewed health as second only to financial services market as a market opportunity.
IBM said it “remains committed to Watson, our broader AI business, and to the clients and partners we support in healthcare IT.”
The company reportedly invested billions of dollars in Watson, but according to a Wall Street Journal report last year, the health business – which provided cloud-based access to the supercomputer and a range of analytics services – has struggled to build market share and reach profitability.
An investigation by Stat meanwhile suggested that Watson Health’s early push into cancer for example was affected by a premature launch, interoperability challenges and over-reliance on human input to generate results.
For its part, IBM has said that the Watson for Oncology product has been improving year-on-year as the AI crunches more and more data.
That is backed up by a meta analysis of its performance published last year in Nature found that the treatment recommendations delivered by the tool were largely in line with human doctors for several cancer types.
However, the study also found that there was less consistency in more advanced cancers, and the authors noted the system “still needs further improvement.”
Watson Health offers a range of other services of course, including
tools for genomic analysis and
running clinical trials that have found favour with a number of pharma companies.
Francisco said in a statement that it offers “a market leading team [that] provides its customers with mission critical products and outstanding service.”
The deal is expected to close in the second quarter, with the current management of Watson Health retaining “similar roles” in the new standalone company, according to the investment company.
IBM’s step back from health comes as tech rivals are still piling into the sector.
@pharma_BI is asking: What will be the future of WATSON Health?
@AVIVA1950 says on 1/26/2022:
Aviva believes plausible scenarios will be that Francisco Partners will:
A. Invest in Watson Health – Like New Mountains Capital (NMC) did with Cytel
B. Acquire several other complementary businesses – Like New Mountains Capital (NMC) did with Cytel
C. Hold and grow – Like New Mountains Capital (NMC) is doing with Cytel since 2018.
D. Sell it in 7 years to @Illumina or @Nvidia or Google’s Parent @AlphaBet
1/21/2022
IBM said Friday it will sell the core data assets of its Watson Health division to a San Francisco-based private equity firm, marking the staggering collapse of its ambitious artificial intelligence effort that failed to live up to its promises to transform everything from drug discovery to cancer care.
IBM has reached an agreement to sell its Watson Health data and analytics business to the private-equity firm Francisco Partners. … He said the deal will give Francisco Partners data and analytics assets that will benefit from “the enhanced investment and expertise of a healthcare industry focused portfolio.”5 days ago
5 days ago — IBM has been trying to find buyers for the Watson Health business for more than a year. And it was seeking a sale price of about $1 billion, The …Missing: Statement | Must include: Statement
5 days ago — IBM Watson Health – Certain Assets Sold: Executive Perspectives. In a prepared statement about the deal, Tom Rosamilia, senior VP, IBM Software, …
Feb 18, 2021 — International Business Machines Corp. is exploring a potential sale of its IBM Watson Health business, according to people familiar with the …
3 days ago — Nuance played a part in building watson in supplying the speech recognition component of Watson. Through the years, Nuance has done some serious …
From: Heidi Rheim et al. GA4GH: International policies and standards for data sharing across genomic research and healthcare. (2021): Cell Genomics, Volume 1 Issue 2.
Siloing genomic data in institutions/jurisdictions limits learning and knowledge
GA4GH policy frameworks enable responsible genomic data sharing
GA4GH technical standards ensure interoperability, broad access, and global benefits
Data sharing across research and healthcare will extend the potential of genomics
Summary
The Global Alliance for Genomics and Health (GA4GH) aims to accelerate biomedical advances by enabling the responsible sharing of clinical and genomic data through both harmonized data aggregation and federated approaches. The decreasing cost of genomic sequencing (along with other genome-wide molecular assays) and increasing evidence of its clinical utility will soon drive the generation of sequence data from tens of millions of humans, with increasing levels of diversity. In this perspective, we present the GA4GH strategies for addressing the major challenges of this data revolution. We describe the GA4GH organization, which is fueled by the development efforts of eight Work Streams and informed by the needs of 24 Driver Projects and other key stakeholders. We present the GA4GH suite of secure, interoperable technical standards and policy frameworks and review the current status of standards, their relevance to key domains of research and clinical care, and future plans of GA4GH. Broad international participation in building, adopting, and deploying GA4GH standards and frameworks will catalyze an unprecedented effort in data sharing that will be critical to advancing genomic medicine and ensuring that all populations can access its benefits.
In order for genomic and personalized medicine to come to fruition it is imperative that data siloes around the world are broken down, allowing the international collaboration for the collection, storage, transferring, accessing and analying of molecular and health-related data.
We had talked on this site in numerous articles about the problems data siloes produce. By data siloes we are meaning that collection and storage of not only DATA but intellectual thought are being held behind physical, electronic, and intellectual walls and inacessible to other scientisits not belonging either to a particular institituion or even a collaborative network.
Standardization and harmonization of data is key to this effort to sharing electronic records. The EU has taken bold action in this matter. The following section is about the General Data Protection Regulation of the EU and can be found at the following link:
The data protection package adopted in May 2016 aims at making Europe fit for the digital age. More than 90% of Europeans say they want the same data protection rights across the EU and regardless of where their data is processed.
The General Data Protection Regulation (GDPR)
Regulation (EU) 2016/679 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data. This text includes the corrigendum published in the OJEU of 23 May 2018.
The regulation is an essential step to strengthen individuals’ fundamental rights in the digital age and facilitate business by clarifying rules for companies and public bodies in the digital single market. A single law will also do away with the current fragmentation in different national systems and unnecessary administrative burdens.
Directive (EU) 2016/680 on the protection of natural persons regarding processing of personal data connected with criminal offences or the execution of criminal penalties, and on the free movement of such data.
The directive protects citizens’ fundamental right to data protection whenever personal data is used by criminal law enforcement authorities for law enforcement purposes. It will in particular ensure that the personal data of victims, witnesses, and suspects of crime are duly protected and will facilitate cross-border cooperation in the fight against crime and terrorism.
The directive entered into force on 5 May 2016 and EU countries had to transpose it into their national law by 6 May 2018.
The following paper by the organiztion The Global Alliance for Genomics and Health discusses these types of collaborative efforts to break down data silos in personalized medicine. This organization has over 2000 subscribers in over 90 countries encompassing over 60 organizations.
Enabling responsible genomic data sharing for the benefit of human health
The Global Alliance for Genomics and Health (GA4GH) is a policy-framing and technical standards-setting organization, seeking to enable responsible genomic data sharing within a human rights framework.
he Global Alliance for Genomics and Health (GA4GH) is an international, nonprofit alliance formed in 2013 to accelerate the potential of research and medicine to advance human health. Bringing together 600+ leading organizations working in healthcare, research, patient advocacy, life science, and information technology, the GA4GH community is working together to create frameworks and standards to enable the responsible, voluntary, and secure sharing of genomic and health-related data. All of our work builds upon the Framework for Responsible Sharing of Genomic and Health-Related Data.
GA4GH Connect is a five-year strategic plan that aims to drive uptake of standards and frameworks for genomic data sharing within the research and healthcare communities in order to enable responsible sharing of clinical-grade genomic data by 2022. GA4GH Connect links our Work Streams with Driver Projects—real-world genomic data initiatives that help guide our development efforts and pilot our tools.
The Global Alliance for Genomics and Health (GA4GH) is a worldwide alliance of genomics researchers, data scientists, healthcare practitioners, and other stakeholders. We are collaborating to establish policy frameworks and technical standards for responsible, international sharing of genomic and other molecular data as well as related health data. Founded in 2013,3 the GA4GH community now consists of more than 1,000 individuals across more than 90 countries working together to enable broad sharing that transcends the boundaries of any single institution or country (see https://www.ga4gh.org).In this perspective, we present the strategic goals of GA4GH and detail current strategies and operational approaches to enable responsible sharing of clinical and genomic data, through both harmonized data aggregation and federated approaches, to advance genomic medicine and research. We describe technical and policy development activities of the eight GA4GH Work Streams and implementation activities across 24 real-world genomic data initiatives (“Driver Projects”). We review how GA4GH is addressing the major areas in which genomics is currently deployed including rare disease, common disease, cancer, and infectious disease. Finally, we describe differences between genomic sequence data that are generated for research versus healthcare purposes, and define strategies for meeting the unique challenges of responsibly enabling access to data acquired in the clinical setting.
GA4GH organization
GA4GH has partnered with 24 real-world genomic data initiatives (Driver Projects) to ensure its standards are fit for purpose and driven by real-world needs. Driver Projects make a commitment to help guide GA4GH development efforts and pilot GA4GH standards (see Table 2). Each Driver Project is expected to dedicate at least two full-time equivalents to GA4GH standards development, which takes place in the context of GA4GH Work Streams (see Figure 1). Work Streams are the key production teams of GA4GH, tackling challenges in eight distinct areas across the data life cycle (see Box 1). Work Streams consist of experts from their respective sub-disciplines and include membership from Driver Projects as well as hundreds of other organizations across the international genomics and health community.
Figure 1Matrix structure of the Global Alliance for Genomics and HealthShow full caption
Box 1GA4GH Work Stream focus areasThe GA4GH Work Streams are the key production teams of the organization. Each tackles a specific area in the data life cycle, as described below (URLs listed in the web resources).
(1)Data use & researcher identities: Develops ontologies and data models to streamline global access to datasets generated in any country9,10
(2)Genomic knowledge standards: Develops specifications and data models for exchanging genomic variant observations and knowledge18
(3)Cloud: Develops federated analysis approaches to support the statistical rigor needed to learn from large datasets
(4)Data privacy & security: Develops guidelines and recommendations to ensure identifiable genomic and phenotypic data remain appropriately secure without sacrificing their analytic potential
(5)Regulatory & ethics: Develops policies and recommendations for ensuring individual-level data are interoperable with existing norms and follow core ethical principles
(6)Discovery: Develops data models and APIs to make data findable, accessible, interoperable, and reusable (FAIR)
(7)Clinical & phenotypic data capture & exchange: Develops data models to ensure genomic data is most impactful through rich metadata collected in a standardized way
(8)Large-scale genomics: Develops APIs and file formats to ensure harmonized technological platforms can support large-scale computing
For more articles on Open Access, Science 2.0, and Data Networks for Genomics on this Open Access Scientific Journal see:
The Vibrant Philly Biotech Scene: Proteovant Therapeutics Using Artificial Intelligence and Machine Learning to Develop PROTACs
Reporter:Stephen J. Williams, Ph.D.
It has been a while since I have added to this series but there have been a plethora of exciting biotech startups in the Philadelphia area, and many new startups combining technology, biotech, and machine learning. One such exciting biotech is Proteovant Therapeutics, which is combining the new PROTAC (Proteolysis-Targeting Chimera) technology with their in house ability to utilize machine learning and artificial intelligence to design these types of compounds to multiple intracellular targets.
PROTACs (which actually is under a trademark name of Arvinus Operations, but is also refered to as Protein Degraders. These PROTACs take advantage of the cell protein homeostatic mechanism of ubiquitin-mediated protein degradation, which is a very specific targeted process which regulates protein levels of various transcription factors, protooncogenes, and receptors. In essence this regulated proteolyic process is needed for normal cellular function, and alterations in this process may lead to oncogenesis, or a proteotoxic crisis leading to mitophagy, autophagy and cellular death. The key to this technology is using chemical linkers to associate an E3 ligase with a protein target of interest. E3 ligases are the rate limiting step in marking the proteins bound for degradation by the proteosome with ubiquitin chains.
A review of this process as well as PROTACs can be found elsewhere in articles (and future articles) on this Open Access Journal.
Protevant have made two important collaborations:
Oncopia Therapeutics: came out of University of Michigan Innovation Hub and lab of Shaomeng Wang, who developed a library of BET and MDM2 based protein degraders. In 2020 was aquired by Riovant Sciences.
Riovant Sciences: uses computer aided design of protein degraders
Proteovant Company Description:
Proteovant is a newly launched development-stage biotech company focusing on discovery and development of disease-modifying therapies by harnessing natural protein homeostasis processes. We have recently acquired numerous assets at discovery and development stages from Oncopia, a protein degradation company. Our lead program is on track to enter IND in 2021. Proteovant is building a strong drug discovery engine by combining deep drugging expertise with innovative platforms including Roivant’s AI capabilities to accelerate discovery and development of protein degraders to address unmet needs across all therapeutic areas. The company has recently secured $200M funding from SK Holdings in addition to investment from Roivant Sciences. Our current therapeutic focus includes but is not limited to oncology, immunology and neurology. We remain agnostic to therapeutic area and will expand therapeutic focus based on opportunity. Proteovant is expanding its discovery and development teams and has multiple positions in biology, chemistry, biochemistry, DMPK, bioinformatics and CMC at many levels. Our R&D organization is located close to major pharmaceutical companies in Eastern Pennsylvania with a second site close to biotech companies in Boston area.
The ubiquitin proteasome system (UPS) is responsible for maintaining protein homeostasis. Targeted protein degradation by the UPS is a cellular process that involves marking proteins and guiding them to the proteasome for destruction. We leverage this physiological cellular machinery to target and destroy disease-causing proteins.
Unlike traditional small molecule inhibitors, our approach is not limited by the classic “active site” requirements. For example, we can target transcription factors and scaffold proteins that lack a catalytic pocket. These classes of proteins, historically, have been very difficult to drug. Further, we selectively degrade target proteins, rather than isozymes or paralogous proteins with high homology. Because of the catalytic nature of the interactions, it is possible to achieve efficacy at lower doses with prolonged duration while decreasing dose-limiting toxicities.
Biological targets once deemed “undruggable” are now within reach.
Roivant develops transformative medicines faster by building technologies and developing talent in creative ways, leveraging the Roivant platform to launch “Vants” – nimble and focused biopharmaceutical and health technology companies. These Vants include Proteovant but also Dermovant, ImmunoVant,as well as others.
Roivant’s drug discovery capabilities include the leading computational physics-based platform for in silico drug design and optimization as well as machine learning-based models for protein degradation.
The integration of our computational and experimental engines enables the rapid design of molecules with high precision and fidelity to address challenging targets for diseases with high unmet need.
Our current modalities include small molecules, heterobifunctionals and molecular glues.
Roivant Unveils Targeted Protein Degradation Platform
– First therapeutic candidate on track to enter clinical studies in 2021
– Computationally-designed degraders for six targets currently in preclinical development
– Acquisition of Oncopia Therapeutics and research collaboration with lab of Dr. Shaomeng Wang at the University of Michigan to add diverse pipeline of current and future compounds
– Clinical-stage degraders will provide foundation for multiple new Vants in distinct disease areas
– Platform supported by $200 million strategic investment from SK Holdings
Other articles in this Vibrant Philly Biotech Scene on this Online Open Access Journal include:
The Map of human proteins drawn by artificial intelligence and PROTAC (proteolysis targeting chimeras) Technology for Drug Discovery
Curators: Dr. Stephen J. Williams and Aviva Lev-Ari, PhD, RN
UPDATED on 11/5/2021
Introducing Isomorphic Labs
I believe we are on the cusp of an incredible new era of biological and medical research. Last year DeepMind’s breakthrough AI system AlphaFold2 was recognised as a solution to the 50-year-old grand challenge of protein folding, capable of predicting the 3D structure of a protein directly from its amino acid sequence to atomic-level accuracy. This has been a watershed moment for computational and AI methods for biology. Building on this advance, today, I’m thrilled to announce the creation of a new Alphabet company – Isomorphic Labs – a commercial venture with the mission to reimagine the entire drug discovery process from the ground up with an AI-first approach and, ultimately, to model and understand some of the fundamental mechanisms of life.
For over a decade DeepMind has been in the vanguard of advancing the state-of-the-art in AI, often using games as a proving ground for developing general purpose learning systems, like AlphaGo, our program that beat the world champion at the complex game of Go. We are at an exciting moment in history now where these techniques and methods are becoming powerful and sophisticated enough to be applied to real-world problems including scientific discovery itself. One of the most important applications of AI that I can think of is in the field of biological and medical research, and it is an area I have been passionate about addressing for many years. Now the time is right to push this forward at pace, and with the dedicated focus and resources that Isomorphic Labs will bring.
An AI-first approach to drug discovery and biology The pandemic has brought to the fore the vital work that brilliant scientists and clinicians do every day to understand and combat disease. We believe that the foundational use of cutting edge computational and AI methods can help scientists take their work to the next level, and massively accelerate the drug discovery process. AI methods will increasingly be used not just for analysing data, but to also build powerful predictive and generative models of complex biological phenomena. AlphaFold2 is an important first proof point of this, but there is so much more to come. At its most fundamental level, I think biology can be thought of as an information processing system, albeit an extraordinarily complex and dynamic one. Taking this perspective implies there may be a common underlying structure between biology and information science – an isomorphic mapping between the two – hence the name of the company. Biology is likely far too complex and messy to ever be encapsulated as a simple set of neat mathematical equations. But just as mathematics turned out to be the right description language for physics, biology may turn out to be the perfect type of regime for the application of AI. What’s next for Isomorphic Labs This is just the beginning of what we hope will become a radical new approach to drug discovery, and I’m incredibly excited to get this ambitious new commercial venture off the ground and to partner with pharmaceutical and biomedical companies. I will serve as CEO for Isomorphic’s initial phase, while remaining as DeepMind CEO, partially to help facilitate collaboration between the two companies where relevant, and to set out the strategy, vision and culture of the new company. This will of course include the building of a world-class multidisciplinary team, with deep expertise in areas such as AI, biology, medicinal chemistry, biophysics, and engineering, brought together in a highly collaborative and innovative environment. (We are hiring!) As pioneers in the emerging field of ‘digital biology’, we look forward to helping usher in an amazingly productive new age of biomedical breakthroughs. Isomorphic’s mission could not be a more important one: to use AI to accelerate drug discovery, and ultimately, find cures for some of humanity’s most devastating diseases.
AI research lab DeepMind has created the most comprehensive map of human proteins to date using artificial intelligence. The company, a subsidiary of Google-parent Alphabet, is releasing the data for free, with some scientists comparing the potential impact of the work to that of the Human Genome Project, an international effort to map every human gene.
Proteins are long, complex molecules that perform numerous tasks in the body, from building tissue to fighting disease. Their purpose is dictated by their structure, which folds like origami into complex and irregular shapes. Understanding how a protein folds helps explain its function, which in turn helps scientists with a range of tasks — from pursuing fundamental research on how the body works, to designing new medicines and treatments. “the culmination of the entire 10-year-plus lifetime of DeepMind” Previously, determining the structure of a protein relied on expensive and time-consuming experiments. But last year DeepMind showed it can produce accurate predictions of a protein’s structure using AI software called AlphaFold. Now, the company is releasing hundreds of thousands of predictions made by the program to the public. “I see this as the culmination of the entire 10-year-plus lifetime of DeepMind,” company CEO and co-founder Demis Hassabis told The Verge. “From the beginning, this is what we set out to do: to make breakthroughs in AI, test that on games like Go and Atari, [and] apply that to real-world problems, to see if we can accelerate scientific breakthroughs and use those to benefit humanity.”
Two examples of protein structures predicted by AlphaFold (in blue) compared with experimental results (in green). Image: DeepMind
There are currently around 180,000 protein structures available in the public domain, each produced by experimental methods and accessible through the Protein Data Bank. DeepMind is releasing predictions for the structure of some 350,000 proteins across 20 different organisms, including animals like mice and fruit flies, and bacteria like E. coli. (There is some overlap between DeepMind’s data and pre-existing protein structures, but exactly how much is difficult to quantify because of the nature of the models.) Most significantly, the release includes predictions for 98 percent of all human proteins, around 20,000 different structures, which are collectively known as the human proteome. It isn’t the first public dataset of human proteins, but it is the most comprehensive and accurate.
If they want, scientists can download the entire human proteome for themselves, says AlphaFold’s technical lead John Jumper. “There is a HumanProteome.zip effectively, I think it’s about 50 gigabytes in size,” Jumper tells The Verge. “You can put it on a flash drive if you want, though it wouldn’t do you much good without a computer for analysis!” “anyone can use it for anything” After launching this first tranche of data, DeepMind plans to keep adding to the store of proteins, which will be maintained by Europe’s flagship life sciences lab, the European Molecular Biology Laboratory (EMBL). By the end of the year, DeepMind hopes to release predictions for 100 million protein structures, a dataset that will be “transformative for our understanding of how life works,” according to Edith Heard, director general of the EMBL. The data will be free in perpetuity for both scientific and commercial researchers, says Hassabis. “Anyone can use it for anything,” the DeepMind CEO noted at a press briefing. “They just need to credit the people involved in the citation.” The benefits of protein folding
Understanding a protein’s structure is useful for scientists across a range of fields. The information can help design new medicines, synthesize novel enzymes that break down waste materials, and create crops that are resistant to viruses or extreme weather. Already, DeepMind’s protein predictions are being used for medical research, including studying the workings of SARS-CoV-2, the virus that causes COVID-19. “it will definitely have a huge impact for the scientific community” New data will speed these efforts, but scientists note it will still take a lot of time to turn this information into real-world results. “I don’t think it’s going to be something that changes the way patients are treated within the year, but it will definitely have a huge impact for the scientific community,” Marcelo C. Sousa, a professor at the University of Colorado’s biochemistry department, told The Verge. Scientists will have to get used to having such information at their fingertips, says DeepMind senior research scientist Kathryn Tunyasuvunakool. “As a biologist, I can confirm we have no playbook for looking at even 20,000 structures, so this [amount of data] is hugely unexpected,” Tunyasuvunakool told The Verge. “To be analyzing hundreds of thousands of structures — it’s crazy.”
Notably, though, DeepMind’s software produces predictions of protein structures rather than experimentally determined models, which means that in some cases further work will be needed to verify the structure. DeepMind says it spent a lot of time building accuracy metrics into its AlphaFold software, which ranks how confident it is for each prediction.
Example protein structures predicted by AlphaFold. Image: DeepMind Predictions of protein structures are still hugely useful, though. Determining a protein’s structure through experimental methods is expensive, time-consuming, and relies on a lot of trial and error. That means even a low-confidence prediction can save scientists years of work by pointing them in the right direction for research. Helen Walden, a professor of structural biology at the University of Glasgow, tells The Verge that DeepMind’s data will “significantly ease” research bottlenecks, but that “the laborious, resource-draining work of doing the biochemistry and biological evaluation of, for example, drug functions” will remain. Sousa, who has previously used data from AlphaFold in his work, says for scientists the impact will be felt immediately. “In our collaboration we had with DeepMind, we had a dataset with a protein sample we’d had for 10 years, and we’d never got to the point of developing a model that fit,” he says. “DeepMind agreed to provide us with a structure, and they were able to solve the problem in 15 minutes after we’d been sitting on it for 10 years.”
Why protein folding is so difficult
Proteins are constructed from chains of amino acids, which come in 20 different varieties in the human body. As any individual protein can be comprised of hundreds of individual amino acids, each of which can fold and twist in different directions, it means a molecule’s final structure has an incredibly large number of possible configurations. One estimate is that the typical protein can be folded in 10^300 ways — that’s a 1 followed by 300 zeroes.
Protein folding has been a “grand challenge” of biology for decades
Because proteins are too small to examine with microscopes, scientists have had to indirectly determine their structure using expensive and complicated methods like nuclear magnetic resonance and X-ray crystallography. The idea of determining the structure of a protein simply by reading a list of its constituent amino acids has been long theorized but difficult to achieve, leading many to describe it as a “grand challenge” of biology. In recent years, though, computational methods — particularly those using artificial intelligence — have suggested such analysis is possible. With these techniques, AI systems are trained on datasets of known protein structures and use this information to create their own predictions.
DeepMind’s AlphaFold software has significantly increased the accuracy of computational protein-folding, as shown by its performance in the CASP competition. Image: DeepMind Many groups have been working on this problem for years, but DeepMind’s deep bench of AI talent and access to computing resources allowed it to accelerate progress dramatically. Last year, the company competed in an international protein-folding competition known as CASP and blew away the competition. Its results were so accurate that computational biologist John Moult, one of CASP’s co-founders, said that “in some sense the problem [of protein folding] is solved.”
DeepMind’s AlphaFold program has been upgraded since last year’s CASP competition and is now 16 times faster. “We can fold an average protein in a matter of minutes, most cases seconds,” says Hassabis.
@@@@@@@
The company also released the underlying code for AlphaFold last week as open-source, allowing others to build on its work in the future.
@@@@@@@
Liam McGuffin, a professor at Reading University who developed some of the UK’s leading protein-folding software, praised the technical brilliance of AlphaFold, but also noted that the program’s success relied on decades of prior research and public data. “DeepMind has vast resources to keep this database up to date and they are better placed to do this than any single academic group,” McGuffin told The Verge. “I think academics would have got there in the end, but it would have been slower because we’re not as well resourced.”
Why does DeepMind care?
Many scientists The Verge spoke to noted the generosity of DeepMind in releasing this data for free. After all, the lab is owned by Google-parent Alphabet, which has been pouring huge amounts of resources into commercial healthcare projects. DeepMind itself loses a lot of money each year, and there have been numerousreportsof tensions between the company and its parent firm over issues like research autonomy and commercial viability.
Hassabis, though, tells The Verge that the company always planned to make this information freely available, and that doing so is a fulfillment of DeepMind’s founding ethos. He stresses that DeepMind’s work is used in lots of places at Google — “almost anything you use, there’s some of our technology that’s part of that under the hood” — but that the company’s primary goal has always been fundamental research. “There’s many ways value can be attained.”
“The agreement when we got acquired is that we are here primarily to advance the state of AGI and AI technologies and then use that to accelerate scientific breakthroughs,” says Hassabis. “[Alphabet] has plenty of divisions focused on making money,” he adds, noting that DeepMind’s focus on research “brings all sorts of benefits, in terms of prestige and goodwill for the scientific community. There’s many ways value can be attained.” Hassabis predicts that AlphaFold is a sign of things to come — a project that shows the huge potential of artificial intelligence to handle messy problems like human biology.
“I think we’re at a really exciting moment,” he says. “In the next decade, we, and others in the AI field, are hoping to produce amazing breakthroughs that will genuinely accelerate solutions to the really big problems we have here on Earth.”
Abstract PROTACs-induced targeted protein degradation has emerged as a novel therapeutic strategy in drug development and attracted the favor of academic institutions, large pharmaceutical enterprises (e.g., AstraZeneca, Bayer, Novartis, Amgen, Pfizer, GlaxoSmithKline, Merck, and Boehringer Ingelheim, etc.), and biotechnology companies. PROTACs opened a new chapter for novel drug development. However, any new technology will face many new problems and challenges. Perspectives on the potential opportunities and challenges of PROTACs will contribute to the research and development of new protein degradation drugs and degrader tools.
Although PROTAC technology has a bright future in drug development, it also has many challenges as follows: (1) Until now, there is only one example of PROTAC reported for an “undruggable” target; (18) more cases are needed to prove the advantages of PROTAC in “undruggable” targets in the future. (2) “Molecular glue”, existing in nature, represents the mechanism of stabilized protein–protein interactions through small molecule modulators of E3 ligases. For instance, auxin, the plant hormone, binds to the ligase SCF-TIR1 to drive recruitment of Aux/IAA proteins and subsequently triggers its degradation. In addition, some small molecules that induce targeted protein degradation through “molecular glue” mode of action have been reported. (21,22) Furthermore, it has been recently reported that some PROTACs may actually achieve target protein degradation via a mechanism that includes “molecular glue” or via “molecular glue” alone. (23) How to distinguish between these two mechanisms and how to combine them to work together is one of the challenges for future research. (3) Since PROTAC acts in a catalytic mode, traditional methods cannot accurately evaluate the pharmacokinetics (PK) and pharmacodynamics (PD) properties of PROTACs. Thus, more studies are urgently needed to establish PK and PD evaluation systems for PROTACs. (4) How to quickly and effectively screen for target protein ligands that can be used in PROTACs, especially those targeting protein–protein interactions, is another challenge. (5) How to understand the degradation activity, selectivity, and possible off-target effects (based on different targets, different cell lines, and different animal models) and how to rationally design PROTACs etc. are still unclear. (6) The human genome encodes more than 600 E3 ubiquitin ligases. However, there are only very few E3 ligases (VHL, CRBN, cIAPs, and MDM2) used in the design of PROTACs. How to expand E3 ubiquitin ligase scope is another challenge faced in this area.
PROTAC technology is rapidly developing, and with the joint efforts of the vast number of scientists in both academia and industry, these problems shall be solved in the near future.
PROTACs have opened a new chapter for the development of new drugs and novel chemical knockdown tools and brought unprecedented opportunities to the industry and academia, which are mainly reflected in the following aspects: (1) Overcoming drug resistance of cancer. In addition to traditional chemotherapy, kinase inhibitors have been developing rapidly in the past 20 years. (12) Although kinase inhibitors are very effective in cancer therapy, patients often develop drug resistance and disease recurrence, consequently. PROTACs showed greater advantages in drug resistant cancers through degrading the whole target protein. For example, ARCC-4 targeting androgen receptor could overcome enzalutamide-resistant prostate cancer (13) and L18I targeting BTK could overcome C481S mutation. (14) (2) Eliminating both the enzymatic and nonenzymatic functions of kinase. Traditional small molecule inhibitors usually inhibit the enzymatic activity of the target, while PROTACs affect not only the enzymatic activity of the protein but also nonenzymatic activity by degrading the entire protein. For example, FAK possesses the kinase dependent enzymatic functions and kinase independent scaffold functions, but regulating the kinase activity does not successfully inhibit all FAK function. In 2018, a highly effective and selective FAK PROTAC reported by Craig M. Crews’ group showed a far superior activity to clinical candidate drug in cell migration and invasion. (15) Therefore, PROTAC can expand the druggable space of the existing targets and regulate proteins that are difficult to control by traditional small molecule inhibitors. (3) Degrade the “undruggable” protein target. At present, only 20–25% of the known protein targets (include kinases, G protein-coupled receptors (GPCRs), nuclear hormone receptors, and iron channels) can be targeted by using conventional drug discovery technologies. (16,17) The proteins that lack catalytic activity and/or have catalytic independent functions are still regarded as “undruggable” targets. The involvement of Signal Transducer and Activator of Transcription 3 (STAT3) in the multiple signaling pathway makes it an attractive therapeutic target; however, the lack of an obviously druggable site on the surface of STAT3 limited the development of STAT3 inhibitors. Thus, there are still no effective drugs directly targeting STAT3 approved by the Food and Drug Administration (FDA). In November 2019, Shaomeng Wang’s group first reported a potent PROTAC targeting STAT3 with potent biological activities in vitro and in vivo. (18) This successful case confirms the key potential of PROTAC technology, especially in the field of “undruggable” targets, such as K-Ras, a tricky tumor target activated by multiple mutations as G12A, G12C, G12D, G12S, G12 V, G13C, and G13D in the clinic. (19) (4) Fast and reversible chemical knockdown strategy in vivo. Traditional genetic protein knockout technologies, zinc-finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), or CRISPR-Cas9, usually have a long cycle, irreversible mode of action, and high cost, which brings a lot of inconvenience for research, especially in nonhuman primates. In addition, these genetic animal models sometimes produce phenotypic misunderstanding due to potential gene compensation or gene mutation. More importantly, the traditional genetic method cannot be used to study the function of embryonic-lethal genes in vivo. Unlike DNA-based protein knockout technology, PROTACs knock down target proteins directly, rather than acting at the genome level, and are suitable for the functional study of embryonic-lethal proteins in adult organisms. In addition, PROTACs provide exquisite temporal control, allowing the knockdown of a target protein at specific time points and enabling the recovery of the target protein after withdrawal of drug treatment. As a new, rapid and reversible chemical knockdown method, PROTAC can be used as an effective supplement to the existing genetic tools. (20)
Peroxisome proliferator-activated receptor (PPAR-gamma) Receptors Activation: PPARγ transrepression for Angiogenesis in Cardiovascular Disease and PPARγ transactivation for Treatment of Diabetes
Patients with type 2 diabetes may soon receive artificial pancreas and a smartphone app assistance
Curator and Reporter: Dr. Premalata Pati, Ph.D., Postdoc
In a brief, randomized crossover investigation, adults with type 2 diabetes and end-stage renal disease who needed dialysis benefited from an artificial pancreas. Tests conducted by the University of Cambridge and Inselspital, University Hospital of Bern, Switzerland, reveal that now the device can help patients safely and effectively monitor their blood sugar levels and reduce the risk of low blood sugar levels.
Diabetes is the most prevalent cause of kidney failure, accounting for just under one-third (30%) of all cases. As the number of people living with type 2 diabetes rises, so does the number of people who require dialysis or a kidney transplant. Kidney failure raises the risk of hypoglycemia and hyperglycemia, or unusually low or high blood sugar levels, which can lead to problems ranging from dizziness to falls and even coma.
Diabetes management in adults with renal failure is difficult for both the patients and the healthcare practitioners. Many components of their therapy, including blood sugar level targets and medications, are poorly understood. Because most oral diabetes drugs are not indicated for these patients, insulin injections are the most often utilized diabetic therapy-yet establishing optimum insulin dose regimes is difficult.
Patients living with type 2 diabetes and kidney failure are a particularly vulnerable group and managing their condition-trying to prevent potentially dangerous highs or lows of blood sugar levels – can be a challenge. There’s a real unmet need for new approaches to help them manage their condition safely and effectively.
The artificial pancreas is a compact, portable medical device that uses digital technology to automate insulin delivery to perform the role of a healthy pancreas in managing blood glucose levels. The system is worn on the outside of the body and consists of three functional components:
a glucose sensor
a computer algorithm for calculating the insulin dose
an insulin pump
The artificial pancreas directed insulin delivery on a Dana Diabecare RS pump using a Dexcom G6 transmitter linked to the Cambridge adaptive model predictive control algorithm, automatically administering faster-acting insulin aspart (Fiasp). The CamDiab CamAPS HX closed-loop app on an unlocked Android phone was used to manage the closed loop system, with a goal glucose of 126 mg/dL. The program calculated an insulin infusion rate based on the data from the G6 sensor every 8 to 12 minutes, which was then wirelessly routed to the insulin pump, with data automatically uploaded to the Diasend/Glooko data management platform.
The Case Study
Between October 2019 and November 2020, the team recruited 26 dialysis patients. Thirteen patients were randomly assigned to get the artificial pancreas first, followed by 13 patients who received normal insulin therapy initially. The researchers compared how long patients spent as outpatients in the target blood sugar range (5.6 to 10.0mmol/L) throughout a 20-day period.
Patients who used the artificial pancreas spent 53 % in the target range on average, compared to 38% who utilized the control treatment. When compared to the control therapy, this translated to approximately 3.5 more hours per day spent in the target range.
The artificial pancreas resulted in reduced mean blood sugar levels (10.1 vs. 11.6 mmol/L). The artificial pancreas cut the amount of time patients spent with potentially dangerously low blood sugar levels, known as ‘hypos.’
The artificial pancreas’ efficacy improved significantly over the research period as the algorithm evolved, and the time spent in the target blood sugar range climbed from 36% on day one to over 60% by the twentieth day. This conclusion emphasizes the need of employing an adaptive algorithm that can adapt to an individual’s fluctuating insulin requirements over time.
When asked if they would recommend the artificial pancreas to others, everyone who responded indicated they would. Nine out of ten (92%) said they spent less time controlling their diabetes with the artificial pancreas than they did during the control period, and a comparable amount (87%) said they were less concerned about their blood sugar levels when using it.
Other advantages of the artificial pancreas mentioned by study participants included fewer finger-prick blood sugar tests, less time spent managing their diabetes, resulting in more personal time and independence, and increased peace of mind and reassurance. One disadvantage was the pain of wearing the insulin pump and carrying the smartphone.
Not only did the artificial pancreas increase the amount of time patients spent within the target range for the blood sugar levels, but it also gave the users peace of mind. They were able to spend less time having to focus on managing their condition and worrying about the blood sugar levels, and more time getting on with their lives.
The team is currently testing the artificial pancreas in outpatient settings in persons with type 2 diabetes who do not require dialysis, as well as in difficult medical scenarios such as perioperative care.
“The artificial pancreas has the potential to become a fundamental part of integrated personalized care for people with complicated medical needs,” said Dr Lia Bally, who co-led the study in Bern.
The authors stated that the study’s shortcomings included a small sample size due to “Brexit-related study funding concerns and the COVID-19 epidemic.”
Boughton concluded:
We would like other clinicians to be aware that automated insulin delivery systems may be a safe and effective treatment option for people with type 2 diabetes and kidney failure in the future.
Science Policy Forum: Should we trust healthcare explanations from AI predictive systems?
Some in industry voice their concerns
Curator: Stephen J. Williams, PhD
Post on AI healthcare and explainable AI
In a Policy Forum article in Science “Beware explanations from AI in health care”, Boris Babic, Sara Gerke, Theodoros Evgeniou, and Glenn Cohen discuss the caveats on relying on explainable versus interpretable artificial intelligence (AI) and Machine Learning (ML) algorithms to make complex health decisions. The FDA has already approved some AI/ML algorithms for analysis of medical images for diagnostic purposes. These have been discussed in prior posts on this site, as well as issues arising from multi-center trials. The authors of this perspective article argue that choice of type of algorithm (explainable versus interpretable) algorithms may have far reaching consequences in health care.
Summary
Artificial intelligence and machine learning (AI/ML) algorithms are increasingly developed in health care for diagnosis and treatment of a variety of medical conditions (1). However, despite the technical prowess of such systems, their adoption has been challenging, and whether and how much they will actually improve health care remains to be seen. A central reason for this is that the effectiveness of AI/ML-based medical devices depends largely on the behavioral characteristics of its users, who, for example, are often vulnerable to well-documented biases or algorithmic aversion (2). Many stakeholders increasingly identify the so-called black-box nature of predictive algorithms as the core source of users’ skepticism, lack of trust, and slow uptake (3, 4). As a result, lawmakers have been moving in the direction of requiring the availability of explanations for black-box algorithmic decisions (5). Indeed, a near-consensus is emerging in favor of explainable AI/ML among academics, governments, and civil society groups. Many are drawn to this approach to harness the accuracy benefits of noninterpretable AI/ML such as deep learning or neural nets while also supporting transparency, trust, and adoption. We argue that this consensus, at least as applied to health care, both overstates the benefits and undercounts the drawbacks of requiring black-box algorithms to be explainable.
Types of AI/ML Algorithms: Explainable and Interpretable algorithms
Interpretable AI: A typical AI/ML task requires constructing algorithms from vector inputs and generating an output related to an outcome (like diagnosing a cardiac event from an image). Generally the algorithm has to be trained on past data with known parameters. When an algorithm is called interpretable, this means that the algorithm uses a transparent or “white box” function which is easily understandable. Such example might be a linear function to determine relationships where parameters are simple and not complex. Although they may not be as accurate as the more complex explainable AI/ML algorithms, they are open, transparent, and easily understood by the operators.
Explainable AI/ML: This type of algorithm depends upon multiple complex parameters and takes a first round of predictions from a “black box” model then uses a second algorithm from an interpretable function to better approximate outputs of the first model. The first algorithm is trained not with original data but based on predictions resembling multiple iterations of computing. Therefore this method is more accurate or deemed more reliable in prediction however is very complex and is not easily understandable. Many medical devices that use an AI/ML algorithm use this type. An example is deep learning and neural networks.
The purpose of both these methodologies is to deal with problems of opacity, or that AI predictions based from a black box undermines trust in the AI.
For a deeper understanding of these two types of algorithms see here:
How interpretability is different from explainability
Why a model might need to be interpretable and/or explainable
Who is working to solve the black box problem—and how
What is interpretability?
Does Chipotle make your stomach hurt? Does loud noise accelerate hearing loss? Are women less aggressive than men? If a machine learning model can create a definition around these relationships, it is interpretable.
All models must start with a hypothesis. Human curiosity propels a being to intuit that one thing relates to another. “Hmm…multiple black people shot by policemen…seemingly out of proportion to other races…something might be systemic?” Explore.
People create internal models to interpret their surroundings. In the field of machine learning, these models can be tested and verified as either accurate or inaccurate representations of the world.
Interpretability means that the cause and effect can be determined.
What is explainability?
ML models are often called black-box models because they allow a pre-set number of empty parameters, or nodes, to be assigned values by the machine learning algorithm. Specifically, the back-propagation step is responsible for updating the weights based on its error function.
To predict when a person might die—the fun gamble one might play when calculating a life insurance premium, and the strange bet a person makes against their own life when purchasing a life insurance package—a model will take in its inputs, and output a percent chance the given person has at living to age 80.
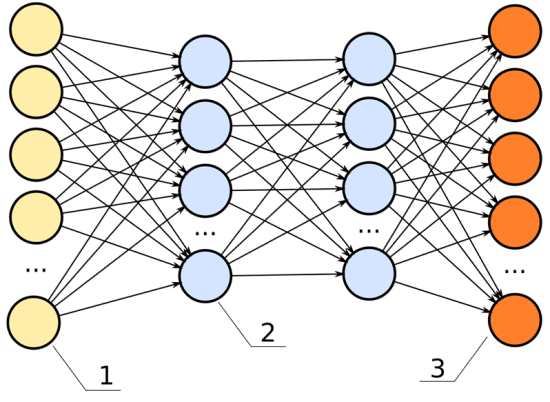
Below is an image of a neural network. The inputs are the yellow; the outputs are the orange. Like a rubric to an overall grade, explainability shows how significant each of the parameters, all the blue nodes, contribute to the final decision.
In this neural network, the hidden layers (the two columns of blue dots) would be the black box.
For example, we have these data inputs:
Age
BMI score
Number of years spent smoking
Career category
If this model had high explainability, we’d be able to say, for instance:
The career category is about 40% important
The number of years spent smoking weighs in at 35% important
The age is 15% important
The BMI score is 10% important
Explainability: important, not always necessary
Explainability becomes significant in the field of machine learning because, often, it is not apparent. Explainability is often unnecessary. A machine learning engineer can build a model without ever having considered the model’s explainability. It is an extra step in the building process—like wearing a seat belt while driving a car. It is unnecessary for the car to perform, but offers insurance when things crash.
The benefit a deep neural net offers to engineers is it creates a black box of parameters, like fake additional data points, that allow a model to base its decisions against. These fake data points go unknown to the engineer. The black box, or hidden layers, allow a model to make associations among the given data points to predict better results. For example, if we are deciding how long someone might have to live, and we use career data as an input, it is possible the model sorts the careers into high- and low-risk career options all on its own.
Perhaps we inspect a node and see it relates oil rig workers, underwater welders, and boat cooks to each other. It is possible the neural net makes connections between the lifespan of these individuals and puts a placeholder in the deep net to associate these. If we were to examine the individual nodes in the black box, we could note this clustering interprets water careers to be a high-risk job.
In the previous chart, each one of the lines connecting from the yellow dot to the blue dot can represent a signal, weighing the importance of that node in determining the overall score of the output.
If that signal is high, that node is significant to the model’s overall performance.
If that signal is low, the node is insignificant.
With this understanding, we can define explainability as:
Knowledge of what one node represents and how important it is to the model’s performance.
So how does choice of these two different algorithms make a difference with respect to health care and medical decision making?
The authors argue:
“Regulators like the FDA should focus on those aspects of the AI/ML system that directly bear on its safety and effectiveness – in particular, how does it perform in the hands of its intended users?”
A suggestion for
Enhanced more involved clinical trials
Provide individuals added flexibility when interacting with a model, for example inputting their own test data
More interaction between user and model generators
Determining in which situations call for interpretable AI versus explainable (for instance predicting which patients will require dialysis after kidney damage)
Other articles on AI/ML in medicine and healthcare on this Open Access Journal include
Al is on the way to lead critical ED decisions on CT
Curator and Reporter: Dr. Premalata Pati, Ph.D., Postdoc
Artificial intelligence (AI) has infiltrated many organizational processes, raising concerns that robotic systems will eventually replace many humans in decision-making. The advent of AI as a tool for improving health care provides new prospects to improve patient and clinical team’s performance, reduce costs, and impact public health. Examples include, but are not limited to, automation; information synthesis for patients, “fRamily” (friends and family unpaid caregivers), and health care professionals; and suggestions and visualization of information for collaborative decision making.
In the emergency department (ED), patients with Crohn’s disease (CD) are routinely subjected to Abdomino-Pelvic Computed Tomography (APCT). It is necessary to diagnose clinically actionable findings (CAF) since they may require immediate intervention, which is typically surgical. Repeated APCTs, on the other hand, results in higher ionizing radiation exposure. The majority of APCT performance guidance is clinical and empiric. Emergency surgeons struggle to identify Crohn’s disease patients who actually require a CT scan to determine the source of acute abdominal distress.
Aid seems to be on the way. Researchers employed machine learning to accurately distinguish these sufferers from Crohn’s patients who appear with the same complaint but may safely avoid the recurrent exposure to contrast materials and ionizing radiation that CT would otherwise wreak on them.
Retrospectively, Jacob Ollech and his fellow researcher have analyzed 101 emergency treatments of patients with Crohn’s who underwent abdominopelvic CT.
They were looking for examples where a scan revealed clinically actionable results. These were classified as intestinal blockage, perforation, intra-abdominal abscess, or complex fistula by the researchers.
On CT, 44 (43.5 %) of the 101 cases reviewed had such findings.
Ollech and colleagues utilized a machine-learning technique to design a decision-support tool that required only four basic clinical factors to test an AI approach for making the call.
The approach was successful in categorizing patients into low- and high-risk groupings. The researchers were able to risk-stratify patients based on the likelihood of clinically actionable findings on abdominopelvic CT as a result of their success.
Ollech and co-authors admit that their limited sample size, retrospective strategy, and lack of external validation are shortcomings.
Moreover, several patients fell into an intermediate risk category, implying that a standard workup would have been required to guide CT decision-making in a real-world situation anyhow.
Consequently, they generate the following conclusion:
We believe this study shows that a machine learning-based tool is a sound approach for better-selecting patients with Crohn’s disease admitted to the ED with acute gastrointestinal complaints about abdominopelvic CT: reducing the number of CTs performed while ensuring that patients with high risk for clinically actionable findings undergo abdominopelvic CT appropriately.
Main Source:
Konikoff, Tom, Idan Goren, Marianna Yalon, Shlomit Tamir, Irit Avni-Biron, Henit Yanai, Iris Dotan, and Jacob E. Ollech. “Machine learning for selecting patients with Crohn’s disease for abdominopelvic computed tomography in the emergency department.” Digestive and Liver Disease (2021). https://www.sciencedirect.com/science/article/abs/pii/S1590865821003340
Other Related Articles published in this Open Access Online Scientific Journal include the following:
From High-Throughput Assay to Systems Biology: New Tools for Drug Discovery
Curator: Stephen J. Williams, PhD
Marc W. Kirschner*
Department of Systems Biology Harvard Medical School
Boston, Massachusetts 02115
With the new excitement about systems biology, there is understandable interest in a definition. This has proven somewhat difficult. Scientific fields, like species, arise by descent with modification, so in their earliest forms even the founders of great dynasties are only marginally different than their sister fields and species. It is only in retrospect that we can recognize the significant founding events. Before embarking on a definition of systems biology, it may be worth remembering that confusion and controversy surrounded the introduction of the term “molecular biology,” with claims that it hardly differed from biochemistry. Yet in retrospect molecular biology was new and different. It introduced both new subject matter and new technological approaches, in addition to a new style.
As a point of departure for systems biology, consider the quintessential experiment in the founding of molecular biology, the one gene one enzyme hypothesis of Beadle and Tatum. This experiment first connected the genotype directly to the phenotype on a molecular level, although efforts in that direction can certainly be found in the work of Archibald Garrod, Sewell Wright, and others. Here a protein (in this case an enzyme) is seen to be a product of a single gene, and a single function; the completion of a specific step in amino acid biosynthesis is the direct result. It took the next 30 years to fill in the gaps in this process. Yet the one gene one enzyme hypothesis looks very different to us today. What is the function of tubulin, of PI-3 kinase or of rac? Could we accurately predict the phenotype of a nonlethal mutation in these genes in a multicellular organism? Although we can connect structure to the gene, we can no longer infer its larger purpose in the cell or in the organism. There are too many purposes; what the protein does is defined by context. The context also includes a history, either developmental or physiological. Thus the behavior of the Wnt signaling pathway depends on the previous lineage, the “where and when” questions of embryonic development. Similarly the behavior of the immune system depends on previous experience in a variable environment. All of these features stress how inadequate an explanation for function we can achieve solely by trying to identify genes (by annotating them!) and characterizing their transcriptional control circuits.
That we are at a crossroads in how to explore biology is not at all clear to many. Biology is hardly in its dotage; the process of discovery seems to have been perfected, accelerated, and made universally applicable to all fields of biology. With the completion of the human genome and the genomes of other species, we have a glimpse of many more genes than we ever had before to study. We are like naturalists discovering a new continent, enthralled with the diversity itself. But we have also at the same time glimpsed the finiteness of this list of genes, a disturbingly small list. We have seen that the diversity of genes cannot approximate the diversity of functions within an organism. In response, we have argued that combinatorial use of small numbers of components can generate all the diversity that is needed. This has had its recent incarnation in the simplistic view that the rules of cis-regulatory control on DNA can directly lead to an understanding of organisms and their evolution. Yet this assumes that the gene products can be linked together in arbitrary combinations, something that is not assured in chemistry. It also downplays the significant regulatory features that involve interactions between gene products, their localization, binding, posttranslational modification, degradation, etc. The big question to understand in biology is not regulatory linkage but the nature of biological systems that allows them to be linked together in many nonlethal and even useful combinations. More and more we come to realize that understanding the conserved genes and their conserved circuits will require an understanding of their special properties that allow them to function together to generate different phenotypes in different tissues of metazoan organisms. These circuits may have certain robustness, but more important they have adaptability and versatility. The ease of putting conserved processes under regulatory control is an inherent design feature of the processes themselves. Among other things it loads the deck in evolutionary variation and makes it more feasible to generate useful phenotypes upon which selection can act.
Systems biology offers an opportunity to study how the phenotype is generated from the genotype and with it a glimpse of how evolution has crafted the phenotype. One aspect of systems biology is the development of techniques to examine broadly the level of protein, RNA, and DNA on a gene by gene basis and even the posttranslational modification and localization of proteins. In a very short time we have witnessed the development of high-throughput biology, forcing us to consider cellular processes in toto. Even though much of the data is noisy and today partially inconsistent and incomplete, this has been a radical shift in the way we tear apart problems one interaction at a time. When coupled with gene deletions by RNAi and classical methods, and with the use of chemical tools tailored to proteins and protein domains, these high-throughput techniques become still more powerful.
High-throughput biology has opened up another important area of systems biology: it has brought us out into the field again or at least made us aware that there is a world outside our laboratories. Our model systems have been chosen intentionally to be of limited genetic diversity and examined in a highly controlled and reproducible environment. The real world of ecology, evolution, and human disease is a very different place. When genetics separated from the rest of biology in the early part of the 20th century, most geneticists sought to understand heredity and chose to study traits in the organism that could be easily scored and could be used to reveal genetic mechanisms. This was later extended to powerful effect to use genetics to study cell biological and developmental mechanisms. Some geneticists, including a large school in Russia in the early 20th century, continued to study the genetics of natural populations, focusing on traits important for survival. That branch of genetics is coming back strongly with the power of phenotypic assays on the RNA and protein level. As human beings we are most concerned not with using our genetic misfortunes to unravel biology’s complexity (important as that is) but with the role of our genetics in our individual survival. The context for understanding this is still not available, even though the data are now coming in torrents, for many of the genes that will contribute to our survival will have small quantitative effects, partially masked or accentuated by other genetic and environmental conditions. To understand the genetic basis of disease will require not just mapping these genes but an understanding of how the phenotype is created in the first place and the messy interactions between genetic variation and environmental variation.
Extracts and explants are relatively accessible to synthetic manipulation. Next there is the explicit reconstruction of circuits within cells or the deliberate modification of those circuits. This has occurred for a while in biology, but the difference is that now we wish to construct or intervene with the explicit purpose of describing the dynamical features of these synthetic or partially synthetic systems. There are more and more tools to intervene and more and more tools to measure. Although these fall short of total descriptions of cells and organisms, the detailed information will give us a sense of the special life-like processes of circuits, proteins, cells in tissues, and whole organisms in their environment. This meso-scale systems biology will help establish the correspondence between molecules and large-scale physiology.
You are probably running out of patience for some definition of systems biology. In any case, I do not think the explicit definition of systems biology should come from me but should await the words of the first great modern systems biologist. She or he is probably among us now. However, if forced to provide some kind of label for systems biology, I would simply say that systems biology is the study of the behavior of complex biological organization and processes in terms of the molecular constituents. It is built on molecular biology in its special concern for information transfer, on physiology for its special concern with adaptive states of the cell and organism, on developmental biology for the importance of defining a succession of physiological states in that process, and on evolutionary biology and ecology for the appreciation that all aspects of the organism are products of selection, a selection we rarely understand on a molecular level. Systems biology attempts all of this through quantitative measurement, modeling, reconstruction, and theory. Systems biology is not a branch of physics but differs from physics in that the primary task is to understand how biology generates variation. No such imperative to create variation exists in the physical world. It is a new principle that Darwin understood and upon which all of life hinges. That sounds different enough for me to justify a new field and a new name. Furthermore, the success of systems biology is essential if we are to understand life; its success is far from assured—a good field for those seeking risk and adventure.
Biologically active small molecules have a central role in drug development, and as chemical probes and tool compounds to perturb and elucidate biological processes. Small molecules can be rationally designed for a given target, or a library of molecules can be screened against a target or phenotype of interest. Especially in the case of phenotypic screening approaches, a major challenge is to translate the compound-induced phenotype into a well-defined cellular target and mode of action of the hit compound. There is no “one size fits all” approach, and recent years have seen an increase in available target deconvolution strategies, rooted in organic chemistry, proteomics, and genetics. This review provides an overview of advances in target identification and mechanism of action studies, describes the strengths and weaknesses of the different approaches, and illustrates the need for chemical biologists to integrate and expand the existing tools to increase the probability of evolving screen hits to robust chemical probes.
5.1.5. Large-Scale Proteomics
While FITExP is based on protein expression regulation during apoptosis, a study of Ruprecht et al. showed that proteomic changes are induced both by cytotoxic and non-cytotoxic compounds, which can be detected by mass spectrometry to give information on a compound’s mechanism of action. They developed a large-scale proteome-wide mass spectrometry analysis platform for MOA studies, profiling five lung cancer cell lines with over 50 drugs. Aggregation analysis over the different cell lines and the different compounds showed that one-quarter of the drugs changed the abundance of their protein target. This approach allowed target confirmation of molecular degraders such as PROTACs or molecular glues. Finally, this method yielded unexpected off-target mechanisms for the MAP2K1/2 inhibitor PD184352 and the ALK inhibitor ceritinib [97]. While such a mapping approach clearly provides a wealth of information, it might not be easily attainable for groups that are not equipped for high-throughput endeavors.
All-in-all, mass spectrometry methods have gained a lot of traction in recent years and have been successfully applied for target deconvolution and MOA studies of small molecules. As with all high-throughput methods, challenges lie in the accessibility of the instruments (both from a time and cost perspective) and data analysis of complex and extensive data sets.
5.2. Genetic Approaches
Both label-based and mass spectrometry proteomic approaches are based on the physical interaction between a small molecule and a protein target, and focus on the proteome for target deconvolution. It has been long realized that genetics provides an alternative avenue to understand a compound’s action, either through precise modification of protein levels, or by inducing protein mutations. First realized in yeast as a genetically tractable organism over 20 years ago, recent advances in genetic manipulation of mammalian cells have opened up important opportunities for target identification and MOA studies through genetic screening in relevant cell types [98]. Genetic approaches can be roughly divided into two main areas, with the first centering on the identification of mutations that confer compound resistance (Figure 3a), and the second on genome-wide perturbation of gene function and the concomitant changes in sensitivity to the compound (Figure 3b). While both methods can be used to identify or confirm drug targets, the latter category often provides many additional insights in the compound’s mode of action.
Figure 3. Genetic methods for target identification and mode of action studies. Schematic representations of (a) resistance cloning, and (b) chemogenetic interaction screens.
5.2.1. Resistance Cloning
The “gold standard” in drug target confirmation is to identify mutations in the presumed target protein that render it insensitive to drug treatment. Conversely, different groups have sought to use this principle as a target identification method based on the concept that cells grown in the presence of a cytotoxic drug will either die or develop mutations that will make them resistant to the compound. With recent advances in deep sequencing it is now possible to then scan the transcriptome [99] or genome [100] of the cells for resistance-inducing mutations. Genes that are mutated are then hypothesized to encode the protein target. For this approach to be successful, there are two initial requirements: (1) the compound needs to be cytotoxic for resistant clones to arise, and (2) the cell line needs to be genetically unstable for mutations to occur in a reasonable timeframe.
In 2012, the Kapoor group demonstrated in a proof-of-concept study that resistance cloning in mammalian cells, coupled to transcriptome sequencing (RNA-seq), yields the known polo-like kinase 1 (PLK1) target of the small molecule BI 2536. For this, they used the cancer cell line HCT-116, which is deficient in mismatch repair and consequently prone to mutations. They generated and sequenced multiple resistant clones, and clustered the clones based on similarity. PLK1 was the only gene that was mutated in multiple groups. Of note, one of the groups did not contain PLK1 mutations, but rather developed resistance through upregulation of ABCBA1, a drug efflux transporter, which is a general and non-specific resistance mechanism [101]. In a following study, they optimized their pipeline “DrugTargetSeqR”, by counter-screening for these types of multidrug resistance mechanisms so that these clones were excluded from further analysis (Figure 3a). Furthermore, they used CRISPR/Cas9-mediated gene editing to determine which mutations were sufficient to confer drug resistance, and as independent validation of the biochemical relevance of the obtained hits [102].
While HCT-116 cells are a useful model cell line for resistance cloning because of their genomic instability, they may not always be the cell line of choice, depending on the compound and process that is studied. Povedana et al. used CRISPR/Cas9 to engineer mismatch repair deficiencies in Ewing sarcoma cells and small cell lung cancer cells. They found that deletion of MSH2 results in hypermutations in these normally mutationally silent cells, resulting in the formation of resistant clones in the presence of bortezomib, MLN4924, and CD437, which are all cytotoxic compounds [103]. Recently, Neggers et al. reasoned that CRISPR/Cas9-induced non-homologous end-joining repair could be a viable strategy to create a wide variety of functional mutants of essential genes through in-frame mutations. Using a tiled sgRNA library targeting 75 target genes of investigational neoplastic drugs in HAP1 and K562 cells, they generated several KPT-9274 (an anticancer agent with unknown target)-resistant clones, and subsequent deep sequencing showed that the resistant clones were enriched in NAMPT sgRNAs. Direct target engagement was confirmed by co-crystallizing the compound with NAMPT [104]. In addition to these genetic mutation strategies, an alternative method is to grow the cells in the presence of a mutagenic chemical to induce higher mutagenesis rates [105,106].
When there is already a hypothesis on the pathway involved in compound action, the resistance cloning methodology can be extended to non-cytotoxic compounds. Sekine et al. developed a fluorescent reporter model for the integrated stress response, and used this cell line for target deconvolution of a small molecule inhibitor towards this pathway (ISRIB). Reporter cells were chemically mutagenized, and ISRIB-resistant clones were isolated by flow cytometry, yielding clones with various mutations in the delta subunit of guanine nucleotide exchange factor eIF2B [107].
While there are certainly successful examples of resistance cloning yielding a compound’s direct target as discussed above, resistance could also be caused by mutations or copy number alterations in downstream components of a signaling pathway. This is illustrated by clinical examples of acquired resistance to small molecules, nature’s way of “resistance cloning”. For example, resistance mechanisms in Hedgehog pathway-driven cancers towards the Smoothened inhibitor vismodegib include compound-resistant mutations in Smoothened, but also copy number changes in downstream activators SUFU and GLI2 [108]. It is, therefore, essential to conduct follow-up studies to confirm a direct interaction between a compound and the hit protein, as well as a lack of interaction with the mutated protein.
5.2.3. “Chemogenomics”: Examples of Gene-Drug Interaction Screens
When genetic perturbations are combined with small molecule drugs in a chemogenetic interaction screen, the effect of a gene’s perturbation on compound action is studied. Gene perturbation can render the cells resistant to the compound (suppressor interaction), or conversely, result in hypersensitivity and enhanced compound potency (synergistic interaction) [5,117,121]. Typically, cells are treated with the compound at a sublethal dose, to ascertain that both types of interactions can be found in the final dataset, and often it is necessary to use a variety of compound doses (i.e., LD20, LD30, LD50) and timepoints to obtain reliable insights (Figure 3b).
An early example of successful coupling of a phenotypic screen and downstream genetic screening for target identification is the study of Matheny et al. They identified STF-118804 as a compound with antileukemic properties. Treatment of MV411 cells, stably transduced with a high complexity, genome-wide shRNA library, with STF-118804 (4 rounds of increasing concentration) or DMSO control resulted in a marked depletion of cells containing shRNAs against nicotinamide phosphoribosyl transferase (NAMPT) [122].
The Bassik lab subsequently directly compared the performance of shRNA-mediated knockdown versus CRISPR/Cas9-knockout screens for the target elucidation of the antiviral drug GSK983. The data coming out of both screens were complementary, with the shRNA screen resulting in hits leading to the direct compound target and the CRISPR screen giving information on cellular mechanisms of action of the compound. A reason for this is likely the level of protein depletion that is reached by these methods: shRNAs lead to decreased protein levels, which is advantageous when studying essential genes. However, knockdown may not result in a phenotype for non-essential genes, in which case a full CRISPR-mediated knockout is necessary to observe effects [123].
Another NAMPT inhibitor was identified in a CRISPR/Cas9 “haplo-insufficiency (HIP)”-like approach [124]. Haploinsuffiency profiling is a well-established system in yeast which is performed in a ~50% protein background by heterozygous deletions [125]. As there is no control over CRISPR-mediated loss of alleles, compound treatment was performed at several timepoints after addition of the sgRNA library to HCT116 cells stably expressing Cas9, in the hope that editing would be incomplete at early timepoints, resulting in residual protein levels. Indeed, NAMPT was found to be the target of phenotypic hit LB-60-OF61, especially at earlier timepoints, confirming the hypothesis that some level of protein needs to be present to identify a compound’s direct target [124]. This approach was confirmed in another study, thereby showing that direct target identification through CRISPR-knockout screens is indeed possible [126].
An alternative strategy was employed by the Weissman lab, where they combined genome-wide CRISPR-interference and -activation screens to identify the target of the phase 3 drug rigosertib. They focused on hits that had opposite action in both screens, as in sensitizing in one but protective in the other, which were related to microtubule stability. In a next step, they created chemical-genetic profiles of a variety of microtubule destabilizing agents, rationalizing that compounds with the same target will have similar drug-gene interactions. For this, they made a focused library of sgRNAs, based on the most high-ranking hits in the rigosertib genome-wide CRISPRi screen, and compared the focused screen results of the different compounds. The profile for rigosertib clustered well with that of ABT-571, and rigorous target validation studies confirmed rigosertib binding to the colchicine binding site of tubulin—the same site as occupied by ABT-571 [127].
From the above examples, it is clear that genetic screens hold a lot of promise for target identification and MOA studies for small molecules. The CRISPR screening field is rapidly evolving, sgRNA libraries are continuously improving and increasingly commercially available, and new tools for data analysis are being developed [128]. The challenge lies in applying these screens to study compounds that are not cytotoxic, where finding the right dosage regimen will not be trivial.
SYSTEMS BIOLOGY AND CANCER RESEARCH & DRUG DISCOVERY
Integrative Analysis of Next-Generation Sequencing for Next-Generation Cancer Research toward Artificial Intelligence
The rapid improvement of next-generation sequencing (NGS) technologies and their application in large-scale cohorts in cancer research led to common challenges of big data. It opened a new research area incorporating systems biology and machine learning. As large-scale NGS data accumulated, sophisticated data analysis methods became indispensable. In addition, NGS data have been integrated with systems biology to build better predictive models to determine the characteristics of tumors and tumor subtypes. Therefore, various machine learning algorithms were introduced to identify underlying biological mechanisms. In this work, we review novel technologies developed for NGS data analysis, and we describe how these computational methodologies integrate systems biology and omics data. Subsequently, we discuss how deep neural networks outperform other approaches, the potential of graph neural networks (GNN) in systems biology, and the limitations in NGS biomedical research. To reflect on the various challenges and corresponding computational solutions, we will discuss the following three topics: (i) molecular characteristics, (ii) tumor heterogeneity, and (iii) drug discovery. We conclude that machine learning and network-based approaches can add valuable insights and build highly accurate models. However, a well-informed choice of learning algorithm and biological network information is crucial for the success of each specific research question
1. Introduction
The development and widespread use of high-throughput technologies founded the era of big data in biology and medicine. In particular, it led to an accumulation of large-scale data sets that opened a vast amount of possible applications for data-driven methodologies. In cancer, these applications range from fundamental research to clinical applications: molecular characteristics of tumors, tumor heterogeneity, drug discovery and potential treatments strategy. Therefore, data-driven bioinformatics research areas have tailored data mining technologies such as systems biology, machine learning, and deep learning, elaborated in this review paper (see Figure 1 and Figure 2). For example, in systems biology, data-driven approaches are applied to identify vital signaling pathways [1]. This pathway-centric analysis is particularly crucial in cancer research to understand the characteristics and heterogeneity of the tumor and tumor subtypes. Consequently, this high-throughput data-based analysis enables us to explore characteristics of cancers with a systems biology and a systems medicine point of view [2].Combining high-throughput techniques, especially next-generation sequencing (NGS), with appropriate analytical tools has allowed researchers to gain a deeper systematic understanding of cancer at various biological levels, most importantly genomics, transcriptomics, and epigenetics [3,4]. Furthermore, more sophisticated analysis tools based on computational modeling are introduced to decipher underlying molecular mechanisms in various cancer types. The increasing size and complexity of the data required the adaptation of bioinformatics processing pipelines for higher efficiency and sophisticated data mining methodologies, particularly for large-scale, NGS datasets [5]. Nowadays, more and more NGS studies integrate a systems biology approach and combine sequencing data with other types of information, for instance, protein family information, pathway, or protein–protein interaction (PPI) networks, in an integrative analysis. Experimentally validated knowledge in systems biology may enhance analysis models and guides them to uncover novel findings. Such integrated analyses have been useful to extract essential information from high-dimensional NGS data [6,7]. In order to deal with the increasing size and complexity, the application of machine learning, and specifically deep learning methodologies, have become state-of-the-art in NGS data analysis.
Figure 1. Next-generation sequencing data can originate from various experimental and technological conditions. Depending on the purpose of the experiment, one or more of the depicted omics types (Genomics, Transcriptomics, Epigenomics, or Single-Cell Omics) are analyzed. These approaches led to an accumulation of large-scale NGS datasets to solve various challenges of cancer research, molecular characterization, tumor heterogeneity, and drug target discovery. For instance, The Cancer Genome Atlas (TCGA) dataset contains multi-omics data from ten-thousands of patients. This dataset facilitates a variety of cancer researches for decades. Additionally, there are also independent tumor datasets, and, frequently, they are analyzed and compared with the TCGA dataset. As the large scale of omics data accumulated, various machine learning techniques are applied, e.g., graph algorithms and deep neural networks, for dimensionality reduction, clustering, or classification. (Created with BioRender.com.)
Figure 2. (a) A multitude of different types of data is produced by next-generation sequencing, for instance, in the fields of genomics, transcriptomics, and epigenomics. (b) Biological networks for biomarker validation: The in vivo or in vitro experiment results are considered ground truth. Statistical analysis on next-generation sequencing data produces candidate genes. Biological networks can validate these candidate genes and highlight the underlying biological mechanisms (Section 2.1). (c) De novo construction of Biological Networks: Machine learning models that aim to reconstruct biological networks can incorporate prior knowledge from different omics data. Subsequently, the model will predict new unknown interactions based on new omics information (Section 2.2). (d) Network-based machine learning: Machine learning models integrating biological networks as prior knowledge to improve predictive performance when applied to different NGS data (Section 2.3). (Created with BioRender.com).
Therefore, a large number of studies integrate NGS data with machine learning and propose a novel data-driven methodology in systems biology [8]. In particular, many network-based machine learning models have been developed to analyze cancer data and help to understand novel mechanisms in cancer development [9,10]. Moreover, deep neural networks (DNN) applied for large-scale data analysis improved the accuracy of computational models for mutation prediction [11,12], molecular subtyping [13,14], and drug repurposing [15,16].
2. Systems Biology in Cancer Research
Genes and their functions have been classified into gene sets based on experimental data. Our understandings of cancer concentrated into cancer hallmarks that define the characteristics of a tumor. This collective knowledge is used for the functional analysis of unseen data.. Furthermore, the regulatory relationships among genes were investigated, and, based on that, a pathway can be composed. In this manner, the accumulation of public high-throughput sequencing data raised many big-data challenges and opened new opportunities and areas of application for computer science. Two of the most vibrantly evolving areas are systems biology and machine learning which tackle different tasks such as understanding the cancer pathways [9], finding crucial genes in pathways [22,53], or predicting functions of unidentified or understudied genes [54]. Essentially, those models include prior knowledge to develop an analysis and enhance interpretability for high-dimensional data [2]. In addition to understanding cancer pathways with in silico analysis, pathway activity analysis incorporating two different types of data, pathways and omics data, is developed to understand heterogeneous characteristics of the tumor and cancer molecular subtyping. Due to its advantage in interpretability, various pathway-oriented methods are introduced and become a useful tool to understand a complex diseases such as cancer [55,56,57].
In this section, we will discuss how two related research fields, namely, systems biology and machine learning, can be integrated with three different approaches (see Figure 2), namely, biological network analysis for biomarker validation, the use of machine learning with systems biology, and network-based models.
2.1. Biological Network Analysis for Biomarker Validation
The detection of potential biomarkers indicative of specific cancer types or subtypes is a frequent goal of NGS data analysis in cancer research. For instance, a variety of bioinformatics tools and machine learning models aim at identify lists of genes that are significantly altered on a genomic, transcriptomic, or epigenomic level in cancer cells. Typically, statistical and machine learning methods are employed to find an optimal set of biomarkers, such as single nucleotide polymorphisms (SNPs), mutations, or differentially expressed genes crucial in cancer progression. Traditionally, resource-intensive in vitro analysis was required to discover or validate those markers. Therefore, systems biology offers in silico solutions to validate such findings using biological pathways or gene ontology information (Figure 2b) [58]. Subsequently, gene set enrichment analysis (GSEA) [50] or gene set analysis (GSA) [59] can be used to evaluate whether these lists of genes are significantly associated with cancer types and their specific characteristics. GSA, for instance, is available via web services like DAVID [60] and g:Profiler [61]. Moreover, other applications use gene ontology directly [62,63]. In addition to gene-set-based analysis, there are other methods that focuse on the topology of biological networks. These approaches evaluate various network structure parameters and analyze the connectivity of two genes or the size and interconnection of their neighbors [64,65]. According to the underlying idea, the mutated gene will show dysfunction and can affect its neighboring genes. Thus, the goal is to find abnormalities in a specific set of genes linked with an edge in a biological network. For instance, KeyPathwayMiner can extract informative network modules in various omics data [66]. In summary, these approaches aim at predicting the effect of dysfunctional genes among neighbors according to their connectivity or distances from specific genes such as hubs [67,68]. During the past few decades, the focus of cancer systems biology extended towards the analysis of cancer-related pathways since those pathways tend to carry more information than a gene set. Such analysis is called Pathway Enrichment Analysis (PEA) [69,70]. The use of PEA incorporates the topology of biological networks. However, simultaneously, the lack of coverage issue in pathway data needs to be considered. Because pathway data does not cover all known genes yet, an integration analysis on omics data can significantly drop in genes when incorporated with pathways. Genes that can not be mapped to any pathway are called ‘pathway orphan.’ In this manner, Rahmati et al. introduced a possible solution to overcome the ‘pathway orphan’ issue [71]. At the bottom line, regardless of whether researchers consider gene-set or pathway-based enrichment analysis, the performance and accuracy of both methods are highly dependent on the quality of the external gene-set and pathway data [72].
2.2. De Novo Construction of Biological Networks
While the known fraction of existing biological networks barely scratches the surface of the whole system of mechanisms occurring in each organism, machine learning models can improve on known network structures and can guide potential new findings [73,74]. This area of research is called de novo network construction (Figure 2c), and its predictive models can accelerate experimental validation by lowering time costs [75,76]. This interplay between in silico biological networks building and mining contributes to expanding our knowledge in a biological system. For instance, a gene co-expression network helps discover gene modules having similar functions [77]. Because gene co-expression networks are based on expressional changes under specific conditions, commonly, inferring a co-expression network requires many samples. The WGCNA package implements a representative model using weighted correlation for network construction that leads the development of the network biology field [78]. Due to NGS developments, the analysis of gene co-expression networks subsequently moved from microarray-based to RNA-seq based experimental data [79]. However, integration of these two types of data remains tricky. Ballouz et al. compared microarray and NGS-based co-expression networks and found the existence of a bias originating from batch effects between the two technologies [80]. Nevertheless, such approaches are suited to find disease-specific co-expressional gene modules. Thus, various studies based on the TCGA cancer co-expression network discovered characteristics of prognostic genes in the network [81]. Accordingly, a gene co-expression network is a condition-specific network rather than a general network for an organism. Gene regulatory networks can be inferred from the gene co-expression network when various data from different conditions in the same organism are available. Additionally, with various NGS applications, we can obtain multi-modal datasets about regulatory elements and their effects, such as epigenomic mechanisms on transcription and chromatin structure. Consequently, a gene regulatory network can consist of solely protein-coding genes or different regulatory node types such as transcription factors, inhibitors, promoter interactions, DNA methylations, and histone modifications affecting the gene expression system [82,83]. More recently, researchers were able to build networks based on a particular experimental setup. For instance, functional genomics or CRISPR technology enables the high-resolution regulatory networks in an organism [84]. Other than gene co-expression or regulatory networks, drug target, and drug repurposing studies are active research areas focusing on the de novo construction of drug-to-target networks to allow the potential repurposing of drugs [76,85].
2.3. Network Based Machine Learning
A network-based machine learning model directly integrates the insights of biological networks within the algorithm (Figure 2d) to ultimately improve predictive performance concerning cancer subtyping or susceptibility to therapy. Following the establishment of high-quality biological networks based on NGS technologies, these biological networks were suited to be integrated into advanced predictive models. In this manner, Zhang et al., categorized network-based machine learning approaches upon their usage into three groups: (i) model-based integration, (ii) pre-processing integration, and (iii) post-analysis integration [7]. Network-based models map the omics data onto a biological network, and proper algorithms travel the network while considering both values of nodes and edges and network topology. In the pre-processing integration, pathway or other network information is commonly processed based on its topological importance. Meanwhile, in the post-analysis integration, omics data is processed solely before integration with a network. Subsequently, omics data and networks are merged and interpreted. The network-based model has advantages in multi-omics integrative analysis. Due to the different sensitivity and coverage of various omics data types, a multi-omics integrative analysis is challenging. However, focusing on gene-level or protein-level information enables a straightforward integration [86,87]. Consequently, when different machine learning approaches tried to integrate two or more different data types to find novel biological insights, one of the solutions is reducing the search space to gene or protein level and integrated heterogeneous datatypes [25,88].
In summary, using network information opens new possibilities for interpretation. However, as mentioned earlier, several challenges remain, such as the coverage issue. Current databases for biological networks do not cover the entire set of genes, transcripts, and interactions. Therefore, the use of networks can lead to loss of information for gene or transcript orphans. The following section will focus on network-based machine learning models and their application in cancer genomics. We will put network-based machine learning into the perspective of the three main areas of application, namely, molecular characterization, tumor heterogeneity analysis, and cancer drug discovery.
3. Network-Based Learning in Cancer Research
As introduced previously, the integration of machine learning with the insights of biological networks (Figure 2d) ultimately aims at improving predictive performance and interpretability concerning cancer subtyping or treatment susceptibility.
3.1. Molecular Characterization with Network Information
Various network-based algorithms are used in genomics and focus on quantifying the impact of genomic alteration. By employing prior knowledge in biological network algorithms, performance compared to non-network models can be improved. A prominent example is HotNet. The algorithm uses a thermodynamics model on a biological network and identifies driver genes, or prognostic genes, in pan-cancer data [89]. Another study introduced a network-based stratification method to integrate somatic alterations and expression signatures with network information [90]. These approaches use network topology and network-propagation-like algorithms. Network propagation presumes that genomic alterations can affect the function of neighboring genes. Two genes will show an exclusive pattern if two genes complement each other, and the function carried by those two genes is essential to an organism [91]. This unique exclusive pattern among genomic alteration is further investigated in cancer-related pathways. Recently, Ku et al. developed network-centric approaches and tackled robustness issues while studying synthetic lethality [92]. Although synthetic lethality was initially discovered in model organisms of genetics, it helps us to understand cancer-specific mutations and their functions in tumor characteristics [91].
Furthermore, in transcriptome research, network information is used to measure pathway activity and its application in cancer subtyping. For instance, when comparing the data of two or more conditions such as cancer types, GSEA as introduced in Section 2 is a useful approach to get an overview of systematic changes [50]. It is typically used at the beginning of a data evaluation [93]. An experimentally validated gene set can provide information about how different conditions affect molecular systems in an organism. In addition to the gene sets, different approaches integrate complex interaction information into GSEA and build network-based models [70]. In contrast to GSEA, pathway activity analysis considers transcriptome data and other omics data and structural information of a biological network. For example, PARADIGM uses pathway topology and integrates various omics in the analysis to infer a patient-specific status of pathways [94]. A benchmark study with pan-cancer data recently reveals that using network structure can show better performance [57]. In conclusion, while the loss of data is due to the incompleteness of biological networks, their integration improved performance and increased interpretability in many cases.
3.2. Tumor Heterogeneity Study with Network Information
The tumor heterogeneity can originate from two directions, clonal heterogeneity and tumor impurity. Clonal heterogeneity covers genomic alterations within the tumor [95]. While de novo mutations accumulate, the tumor obtains genomic alterations with an exclusive pattern. When these genomic alterations are projected on the pathway, it is possible to observe exclusive relationships among disease-related genes. For instance, the CoMEt and MEMo algorithms examine mutual exclusivity on protein–protein interaction networks [96,97]. Moreover, the relationship between genes can be essential for an organism. Therefore, models analyzing such alterations integrate network-based analysis [98].
In contrast, tumor purity is dependent on the tumor microenvironment, including immune-cell infiltration and stromal cells [99]. In tumor microenvironment studies, network-based models are applied, for instance, to find immune-related gene modules. Although the importance of the interaction between tumors and immune cells is well known, detailed mechanisms are still unclear. Thus, many recent NGS studies employ network-based models to investigate the underlying mechanism in tumor and immune reactions. For example, McGrail et al. identified a relationship between the DNA damage response protein and immune cell infiltration in cancer. The analysis is based on curated interaction pairs in a protein–protein interaction network [100]. Most recently, Darzi et al. discovered a prognostic gene module related to immune cell infiltration by using network-centric approaches [101]. Tu et al. presented a network-centric model for mining subnetworks of genes other than immune cell infiltration by considering tumor purity [102].
3.3. Drug Target Identification with Network Information
In drug target studies, network biology is integrated into pharmacology [103]. For instance, Yamanishi et al. developed novel computational methods to investigate the pharmacological space by integrating a drug-target protein network with genomics and chemical information. The proposed approaches investigated such drug-target network information to identify potential novel drug targets [104]. Since then, the field has continued to develop methods to study drug target and drug response integrating networks with chemical and multi-omic datasets. In a recent survey study by Chen et al., the authors compared 13 computational methods for drug response prediction. It turned out that gene expression profiles are crucial information for drug response prediction [105].
Moreover, drug-target studies are often extended to drug-repurposing studies. In cancer research, drug-repurposing studies aim to find novel interactions between non-cancer drugs and molecular features in cancer. Drug-repurposing (or repositioning) studies apply computational approaches and pathway-based models and aim at discovering potential new cancer drugs with a higher probability than de novo drug design [16,106]. Specifically, drug-repurposing studies can consider various areas of cancer research, such as tumor heterogeneity and synthetic lethality. As an example, Lee et al. found clinically relevant synthetic lethality interactions by integrating multiple screening NGS datasets [107]. This synthetic lethality and related-drug datasets can be integrated for an effective combination of anticancer therapeutic strategy with non-cancer drug repurposing.
4. Deep Learning in Cancer Research
DNN models develop rapidly and become more sophisticated. They have been frequently used in all areas of biomedical research. Initially, its development was facilitated by large-scale imaging and video data. While most data sets in the biomedical field would not typically be considered big data, the rapid data accumulation enabled by NGS made it suitable for the application of DNN models requiring a large amount of training data [108]. For instance, in 2019, Samiei et al. used TCGA-based large-scale cancer data as benchmark datasets for bioinformatics machine learning research such as Image-Net in the computer vision field [109]. Subsequently, large-scale public cancer data sets such as TCGA encouraged the wide usage of DNNs in the cancer domain [110]. Over the last decade, these state-of-the-art machine learning methods have been incorporated in many different biological questions [111].
In addition to public cancer databases such as TCGA, the genetic information of normal tissues is stored in well-curated databases such as GTEx [112] and 1000Genomes [113]. These databases are frequently used as control or baseline training data for deep learning [114]. Moreover, other non-curated large-scale data sources such as GEO (https://www.ncbi.nlm.nih.gov/geo/, accessed on 20 May 2021) can be leveraged to tackle critical aspects in cancer research. They store a large-scale of biological data produced under various experimental setups (Figure 1). Therefore, an integration of GEO data and other data requires careful preprocessing. Overall, an increasing amount of datasets facilitate the development of current deep learning in bioinformatics research [115].
4.1. Challenges for Deep Learning in Cancer Research
Many studies in biology and medicine used NGS and produced large amounts of data during the past few decades, moving the field to the big data era. Nevertheless, researchers still face a lack of data in particular when investigating rare diseases or disease states. Researchers have developed a manifold of potential solutions to overcome this lack of data challenges, such as imputation, augmentation, and transfer learning (Figure 3b). Data imputation aims at handling data sets with missing values [116]. It has been studied on various NGS omics data types to recover missing information [117]. It is known that gene expression levels can be altered by different regulatory elements, such as DNA-binding proteins, epigenomic modifications, and post-transcriptional modifications. Therefore, various models integrating such regulatory schemes have been introduced to impute missing omics data [118,119]. Some DNN-based models aim to predict gene expression changes based on genomics or epigenomics alteration. For instance, TDimpute aims at generating missing RNA-seq data by training a DNN on methylation data. They used TCGA and TARGET (https://ocg.cancer.gov/programs/target/data-matrix, accessed on 20 May 2021) data as proof of concept of the applicability of DNN for data imputation in a multi-omics integration study [120]. Because this integrative model can exploit information in different levels of regulatory mechanisms, it can build a more detailed model and achieve better performance than a model build on a single-omics dataset [117,121]. The generative adversarial network (GAN) is a DNN structure for generating simulated data that is different from the original data but shows the same characteristics [122]. GANs can impute missing omics data from other multi-omics sources. Recently, the GAN algorithm is getting more attention in single-cell transcriptomics because it has been recognized as a complementary technique to overcome the limitation of scRNA-seq [123]. In contrast to data imputation and generation, other machine learning approaches aim to cope with a limited dataset in different ways. Transfer learning or few-shot learning, for instance, aims to reduce the search space with similar but unrelated datasets and guide the model to solve a specific set of problems [124]. These approaches train models with data of similar characteristics and types but different data to the problem set. After pre-training the model, it can be fine-tuned with the dataset of interest [125,126]. Thus, researchers are trying to introduce few-shot learning models and meta-learning approaches to omics and translational medicine. For example, Select-ProtoNet applied the ProtoTypical Network [127] model to TCGA transcriptome data and classified patients into two groups according to their clinical status [128]. AffinityNet predicts kidney and uterus cancer subtypes with gene expression profiles [129].
Figure 3. (a) In various studies, NGS data transformed into different forms. The 2-D transformed form is for the convolution layer. Omics data is transformed into pathway level, GO enrichment score, or Functional spectra. (b) DNN application on different ways to handle lack of data. Imputation for missing data in multi-omics datasets. GAN for data imputation and in silico data simulation. Transfer learning pre-trained the model with other datasets and fine-tune. (c) Various types of information in biology. (d) Graph neural network examples. GCN is applied to aggregate neighbor information. (Created with BioRender.com).
4.2. Molecular Charactization with Network and DNN Model
DNNs have been applied in multiple areas of cancer research. For instance, a DNN model trained on TCGA cancer data can aid molecular characterization by identifying cancer driver genes. At the very early stage, Yuan et al. build DeepGene, a cancer-type classifier. They implemented data sparsity reduction methods and trained the DNN model with somatic point mutations [130]. Lyu et al. [131] and DeepGx [132] embedded a 1-D gene expression profile to a 2-D array by chromosome order to implement the convolution layer (Figure 3a). Other algorithms, such as the deepDriver, use k-nearest neighbors for the convolution layer. A predefined number of neighboring gene mutation profiles was the input for the convolution layer. It employed this convolution layer in a DNN by aggregating mutation information of the k-nearest neighboring genes [11]. Instead of embedding to a 2-D image, DeepCC transformed gene expression data into functional spectra. The resulting model was able to capture molecular characteristics by training cancer subtypes [14].
Another DNN model was trained to infer the origin of tissue from single-nucleotide variant (SNV) information of metastatic tumor. The authors built a model by using the TCGA/ICGC data and analyzed SNV patterns and corresponding pathways to predict the origin of cancer. They discovered that metastatic tumors retained their original cancer’s signature mutation pattern. In this context, their DNN model obtained even better accuracy than a random forest model [133] and, even more important, better accuracy than human pathologists [12].
4.3. Tumor Heterogeneity with Network and DNN Model
As described in Section 4.1, there are several issues because of cancer heterogeneity, e.g., tumor microenvironment. Thus, there are only a few applications of DNN in intratumoral heterogeneity research. For instance, Menden et al. developed ’Scaden’ to deconvolve cell types in bulk-cell sequencing data. ’Scaden’ is a DNN model for the investigation of intratumor heterogeneity. To overcome the lack of training datasets, researchers need to generate in silico simulated bulk-cell sequencing data based on single-cell sequencing data [134]. It is presumed that deconvolving cell types can be achieved by knowing all possible expressional profiles of the cell [36]. However, this information is typically not available. Recently, to tackle this problem, single-cell sequencing-based studies were conducted. Because of technical limitations, we need to handle lots of missing data, noises, and batch effects in single-cell sequencing data [135]. Thus, various machine learning methods were developed to process single-cell sequencing data. They aim at mapping single-cell data onto the latent space. For example, scDeepCluster implemented an autoencoder and trained it on gene-expression levels from single-cell sequencing. During the training phase, the encoder and decoder work as denoiser. At the same time, they can embed high-dimensional gene-expression profiles to lower-dimensional vectors [136]. This autoencoder-based method can produce biologically meaningful feature vectors in various contexts, from tissue cell types [137] to different cancer types [138,139].
4.4. Drug Target Identification with Networks and DNN Models
In addition to NGS datasets, large-scale anticancer drug assays enabled the training train of DNNs. Moreover, non-cancer drug response assay datasets can also be incorporated with cancer genomic data. In cancer research, a multidisciplinary approach was widely applied for repurposing non-oncology drugs to cancer treatment. This drug repurposing is faster than de novo drug discovery. Furthermore, combination therapy with a non-oncology drug can be beneficial to overcome the heterogeneous properties of tumors [85]. The deepDR algorithm integrated ten drug-related networks and trained deep autoencoders. It used a random-walk-based algorithm to represent graph information into feature vectors. This approach integrated network analysis with a DNN model validated with an independent drug-disease dataset [15].
The authors of CDRscan did an integrative analysis of cell-line-based assay datasets and other drug and genomics datasets. It shows that DNN models can enhance the computational model for improved drug sensitivity predictions [140]. Additionally, similar to previous network-based models, the multi-omics application of drug-targeted DNN studies can show higher prediction accuracy than the single-omics method. MOLI integrated genomic data and transcriptomic data to predict the drug responses of TCGA patients [141].
4.5. Graph Neural Network Model
In general, the advantage of using a biological network is that it can produce more comprehensive and interpretable results from high-dimensional omics data. Furthermore, in an integrative multi-omics data analysis, network-based integration can improve interpretability over traditional approaches. Instead of pre-/post-integration of a network, recently developed graph neural networks use biological networks as the base structure for the learning network itself. For instance, various pathways or interactome information can be integrated as a learning structure of a DNN and can be aggregated as heterogeneous information. In a GNN study, a convolution process can be done on the provided network structure of data. Therefore, the convolution on a biological network made it possible for the GNN to focus on the relationship among neighbor genes. In the graph convolution layer, the convolution process integrates information of neighbor genes and learns topological information (Figure 3d). Consequently, this model can aggregate information from far-distant neighbors, and thus can outperform other machine learning models [142].
In the context of the inference problem of gene expression, the main question is whether the gene expression level can be explained by aggregating the neighboring genes. A single gene inference study by Dutil et al. showed that the GNN model outperformed other DNN models [143]. Moreover, in cancer research, such GNN models can identify cancer-related genes with better performance than other network-based models, such as HotNet2 and MutSigCV [144]. A recent GNN study with a multi-omics integrative analysis identified 165 new cancer genes as an interactive partner for known cancer genes [145]. Additionally, in the synthetic lethality area, dual-dropout GNN outperformed previous bioinformatics tools for predicting synthetic lethality in tumors [146]. GNNs were also able to classify cancer subtypes based on pathway activity measures with RNA-seq data. Lee et al. implemented a GNN for cancer subtyping and tested five cancer types. Thus, the informative pathway was selected and used for subtype classification [147]. Furthermore, GNNs are also getting more attention in drug repositioning studies. As described in Section 3.3, drug discovery requires integrating various networks in both chemical and genomic spaces (Figure 3d). Chemical structures, protein structures, pathways, and other multi-omics data were used in drug-target identification and repurposing studies (Figure 3c). Each of the proposed applications has a specialty in the different purposes of drug-related tasks. Sun et al. summarized GNN-based drug discovery studies and categorized them into four classes: molecular property and activity prediction, interaction prediction, synthesis prediction, and de novo drug design. The authors also point out four challenges in the GNN-mediated drug discovery. At first, as we described before, there is a lack of drug-related datasets. Secondly, the current GNN models can not fully represent 3-D structures of chemical molecules and protein structures. The third challenge is integrating heterogeneous network information. Drug discovery usually requires a multi-modal integrative analysis with various networks, and GNNs can improve this integrative analysis. Lastly, although GNNs use graphs, stacked layers still make it hard to interpret the model [148].
4.6. Shortcomings in AI and Revisiting Validity of Biological Networks as Prior Knowledge
The previous sections reviewed a variety of DNN-based approaches that present a good performance on numerous applications. However, it is hardly a panacea for all research questions. In the following, we will discuss potential limitations of the DNN models. In general, DNN models with NGS data have two significant issues: (i) data requirements and (ii) interpretability. Usually, deep learning needs a large proportion of training data for reasonable performance which is more difficult to achieve in biomedical omics data compared to, for instance, image data. Today, there are not many NGS datasets that are well-curated and -annotated for deep learning. This can be an answer to the question of why most DNN studies are in cancer research [110,149]. Moreover, the deep learning models are hard to interpret and are typically considered as black-boxes. Highly stacked layers in the deep learning model make it hard to interpret its decision-making rationale. Although the methodology to understand and interpret deep learning models has been improved, the ambiguity in the DNN models’ decision-making hindered the transition between the deep learning model and translational medicine [149,150].
As described before, biological networks are employed in various computational analyses for cancer research. The studies applying DNNs demonstrated many different approaches to use prior knowledge for systematic analyses. Before discussing GNN application, the validity of biological networks in a DNN model needs to be shown. The LINCS program analyzed data of ’The Connectivity Map (CMap) project’ to understand the regulatory mechanism in gene expression by inferring the whole gene expression profiles from a small set of genes (https://lincsproject.org/, accessed on 20 May 2021) [151,152]. This LINCS program found that the gene expression level is inferrable with only nearly 1000 genes. They called this gene list ’landmark genes’. Subsequently, Chen et al. started with these 978 landmark genes and tried to predict other gene expression levels with DNN models. Integrating public large-scale NGS data showed better performance than the linear regression model. The authors conclude that the performance advantage originates from the DNN’s ability to model non-linear relationships between genes [153].
Following this study, Beltin et al. extensively investigated various biological networks in the same context of the inference of gene expression level. They set up a simplified representation of gene expression status and tried to solve a binary classification task. To show the relevance of a biological network, they compared various gene expression levels inferred from a different set of genes, neighboring genes in PPI, random genes, and all genes. However, in the study incorporating TCGA and GTEx datasets, the random network model outperformed the model build on a known biological network, such as StringDB [154]. While network-based approaches can add valuable insights to analysis, this study shows that it cannot be seen as the panacea, and a careful evaluation is required for each data set and task. In particular, this result may not represent biological complexity because of the oversimplified problem setup, which did not consider the relative gene-expressional changes. Additionally, the incorporated biological networks may not be suitable for inferring gene expression profiles because they consist of expression-regulating interactions, non-expression-regulating interactions, and various in vivo and in vitro interactions.
“ However, although recently sophisticated applications of deep learning showed improved accuracy, it does not reflect a general advancement. Depending on the type of NGS data, the experimental design, and the question to be answered, a proper approach and specific deep learning algorithms need to be considered. Deep learning is not a panacea. In general, to employ machine learning and systems biology methodology for a specific type of NGS data, a certain experimental design, a particular research question, the technology, and network data have to be chosen carefully.”
Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
Hutter, C.; Zenklusen, J.C. The cancer genome atlas: Creating lasting value beyond its data. Cell2018, 173, 283–285. [Google Scholar] [CrossRef]
Chuang, H.Y.; Lee, E.; Liu, Y.T.; Lee, D.; Ideker, T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol.2007, 3, 140. [Google Scholar] [CrossRef]
Zhang, W.; Chien, J.; Yong, J.; Kuang, R. Network-based machine learning and graph theory algorithms for precision oncology. NPJ Precis. Oncol.2017, 1, 25. [Google Scholar] [CrossRef] [PubMed]
Ngiam, K.Y.; Khor, W. Big data and machine learning algorithms for health-care delivery. Lancet Oncol.2019, 20, e262–e273. [Google Scholar] [CrossRef]
Creixell, P.; Reimand, J.; Haider, S.; Wu, G.; Shibata, T.; Vazquez, M.; Mustonen, V.; Gonzalez-Perez, A.; Pearson, J.; Sander, C.; et al. Pathway and network analysis of cancer genomes. Nat. Methods2015, 12, 615. [Google Scholar]
Reyna, M.A.; Haan, D.; Paczkowska, M.; Verbeke, L.P.; Vazquez, M.; Kahraman, A.; Pulido-Tamayo, S.; Barenboim, J.; Wadi, L.; Dhingra, P.; et al. Pathway and network analysis of more than 2500 whole cancer genomes. Nat. Commun.2020, 11, 729. [Google Scholar] [CrossRef]
Luo, P.; Ding, Y.; Lei, X.; Wu, F.X. deepDriver: Predicting cancer driver genes based on somatic mutations using deep convolutional neural networks. Front. Genet.2019, 10, 13. [Google Scholar] [CrossRef]
Jiao, W.; Atwal, G.; Polak, P.; Karlic, R.; Cuppen, E.; Danyi, A.; De Ridder, J.; van Herpen, C.; Lolkema, M.P.; Steeghs, N.; et al. A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns. Nat. Commun.2020, 11, 728. [Google Scholar] [CrossRef]
Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin. Cancer Res.2018, 24, 1248–1259. [Google Scholar] [CrossRef]
Gao, F.; Wang, W.; Tan, M.; Zhu, L.; Zhang, Y.; Fessler, E.; Vermeulen, L.; Wang, X. DeepCC: A novel deep learning-based framework for cancer molecular subtype classification. Oncogenesis2019, 8, 44. [Google Scholar] [CrossRef]
Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinformatics2019, 35, 5191–5198. [Google Scholar] [CrossRef]
Issa, N.T.; Stathias, V.; Schürer, S.; Dakshanamurthy, S. Machine and deep learning approaches for cancer drug repurposing. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature2020, 578, 82. [Google Scholar] [CrossRef] [PubMed]
King, M.C.; Marks, J.H.; Mandell, J.B. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol.2010, 28, 1075. [Google Scholar] [CrossRef] [PubMed]
Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol.2009, 27, 1160. [Google Scholar] [CrossRef]
Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol.2014, 5, 412. [Google Scholar] [CrossRef] [PubMed]
Zhao, L.; Lee, V.H.; Ng, M.K.; Yan, H.; Bijlsma, M.F. Molecular subtyping of cancer: Current status and moving toward clinical applications. Brief. Bioinform.2019, 20, 572–584. [Google Scholar] [CrossRef] [PubMed]
Jones, P.A.; Issa, J.P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet.2016, 17, 630. [Google Scholar] [CrossRef] [PubMed]
Huang, S.; Chaudhary, K.; Garmire, L.X. More is better: Recent progress in multi-omics data integration methods. Front. Genet.2017, 8, 84. [Google Scholar] [CrossRef]
Chin, L.; Andersen, J.N.; Futreal, P.A. Cancer genomics: From discovery science to personalized medicine. Nat. Med.2011, 17, 297. [Google Scholar] [CrossRef] [PubMed]
Use of Systems Biology in Anti-Microbial Drug Development
Genomics, Computational Biology and Drug Discovery for Mycobacterial Infections: Fighting the Emergence of Resistance. Asma Munir, Sundeep Chaitanya Vedithi, Amanda K. Chaplin and Tom L. Blundell. Front. Genet., 04 September 2020 | https://doi.org/10.3389/fgene.2020.00965
In an earlier review article (Waman et al., 2019), we discussed various computational approaches and experimental strategies for drug target identification and structure-guided drug discovery. In this review we discuss the impact of the era of precision medicine, where the genome sequences of pathogens can give clues about the choice of existing drugs, and repurposing of others. Our focus is directed toward combatting antimicrobial drug resistance with emphasis on tuberculosis and leprosy. We describe structure-guided approaches to understanding the impacts of mutations that give rise to antimycobacterial resistance and the use of this information in the design of new medicines.
Genome Sequences and Proteomic Structural Databases
In recent years, there have been many focused efforts to define the amino-acid sequences of the M. tuberculosis pan-genome and then to define the three-dimensional structures and functional interactions of these gene products. This work has led to essential genes of the bacteria being revealed and to a better understanding of the genetic diversity in different strains that might lead to a selective advantage (Coll et al., 2018). This will help with our understanding of the mode of antibiotic resistance within these strains and aid structure-guided drug discovery. However, only ∼10% of the ∼4128 proteins have structures determined experimentally.
Several databases have been developed to integrate the genomic and/or structural information linked to drug resistance in Mycobacteria (Table 1). These invaluable resources can contribute to better understanding of molecular mechanisms involved in drug resistance and improvement in the selection of potential drug targets.
There is a dearth of information related to structural aspects of proteins from M. leprae and their oligomeric and hetero-oligomeric organization, which has limited the understanding of physiological processes of the bacillus. The structures of only 12 proteins have been solved and deposited in the protein data bank (PDB). However, the high sequence similarity in protein coding genes between M. leprae and M. tuberculosis allows computational methods to be used for comparative modeling of the proteins of M. leprae. Mainly monomeric models using single template modeling have been defined and deposited in the Swiss Model repository (Bienert et al., 2017), in Modbase (Pieper et al., 2014), and in a collection with other infectious disease agents (Sosa et al., 2018). There is a need for multi-template modeling and building homo- and hetero-oligomeric complexes to better understand the interfaces, druggability and impacts of mutations.
We are now exploiting Vivace, a multi-template modeling pipeline developed in our lab for modeling the proteomes of M. tuberculosis (CHOPIN, see above) and M. abscessus [Mabellini Database (Skwark et al., 2019)], to model the proteome of M. leprae. We emphasize the need for understanding the protein interfaces that are critical to function. An example of this is that of the RNA-polymerase holoenzyme complex from M. leprae. We first modeled the structure of this hetero-hexamer complex and later deciphered the binding patterns of rifampin (Vedithi et al., 2018; Figures 1A,B). Rifampin is a known drug to treat tuberculosis and leprosy. Owing to high rifampin resistance in tuberculosis and emerging resistance in leprosy, we used an approach known as “Computational Saturation Mutagenesis”, to identify sites on the protein that are less impacted by mutations. In this study, we were able to understand the association between predicted impacts of mutations on the structure and phenotypic rifampin-resistance outcomes in leprosy.
FIGURE 2
Figure 2.(A) Stability changes predicted by mCSM for systematic mutations in the ß-subunit of RNA polymerase in M. leprae. The maximum destabilizing effect from among all 19 possible mutations at each residue position is considered as a weighting factor for the color map that gradients from red (high destabilizing effects) to white (neutral to stabilizing effects) (Vedithi et al., 2020). (B) One of the known mutations in the ß-subunit of RNA polymerase, the S437H substitution which resulted in a maximum destabilizing effect [-1.701 kcal/mol (mCSM)] among all 19 possibilities this position. In the mutant, histidine (residue in green) forms hydrogen bonds with S434 and Q438, aromatic interactions with F431, and other ring-ring and π interactions with the surrounding residues which can impact the shape of the rifampin binding pocket and rifampin affinity to the ß-subunit [-0.826 log(affinity fold change) (mCSM-lig)]. Orange dotted lines represent weak hydrogen bond interactions. Ring-ring and intergroup interactions are depicted in cyan. Aromatic interactions are represented in sky-blue and carbonyl interactions in pink dotted lines. Green dotted lines represent hydrophobic interactions (Vedithi et al., 2020).
Examples of Understanding and Combatting Resistance
The availability of whole genome sequences in the present era has greatly enhanced the understanding of emergence of drug resistance in infectious diseases like tuberculosis. The data generated by the whole genome sequencing of clinical isolates can be screened for the presence of drug-resistant mutations. A preliminary in silico analysis of mutations can then be used to prioritize experimental work to identify the nature of these mutations.
FIGURE 3
Figure 3.(A) Mechanism of isoniazid activation and INH-NAD adduct formation. (B) Mutations mapped (Munir et al., 2019) on the structure of KatG (PDB ID:1SJ2; Bertrand et al., 2004).
Other articles related to Computational Biology, Systems Biology, and Bioinformatics on this online journal include: